
Currently viewing the category:
"Séquençage"
 A moitié scientifique et à moitié homme d’affaire, Craig Venter qui n’a pas très bon goût concernant les couvertures de ses livres essaie de mettre la main sur les données de plusieurs centaines de milliers à plusieurs millions de génomes (séquences totales ou profils génétiques). Mais que l’on se rassure c’est pour le bien de l’humanité ou au moins de la transhumanité !
A moitié scientifique et à moitié homme d’affaire, Craig Venter qui n’a pas très bon goût concernant les couvertures de ses livres essaie de mettre la main sur les données de plusieurs centaines de milliers à plusieurs millions de génomes (séquences totales ou profils génétiques). Mais que l’on se rassure c’est pour le bien de l’humanité ou au moins de la transhumanité !
Depuis 2005, les technologies de séquençage n’ont cessé d’être plus rapides et moins chères. En 2014, plus de 225.000 génomes humains étaient déjà séquencés grâce à plusieurs initiatives dont le fameux « 100 000 Genomes Project » britannique lancé en 2013. Début 2014 Illumina lançait une campagne de publicité mettant en scène le HiSeqX Ten, le premier séquenceur permettant d’atteindre la promesse d’un coût de séquençage humain à 1000 $. Cette année AstraZeneca annonçait sa collaboration avec le Human Longevity Institute de Craig Venter permettant à ce dernier un accès aux génomes ou profils génomiques de 2 000 000 de personnes d’ici 2020. En utilisant la seule séquence d’ADN, Venter dit que son entreprise peut maintenant prédire la taille, le poids, la couleur des yeux et la couleur des cheveux d’une personne, et produire une image approximative de son visage. Une grande partie de ces « détails » est dissimulé dans les variations rares, dit Venter, dont le propre génome a été mis à disposition dans les bases de données publiques depuis plus d’une décennie. Soit dit en passant, même ce promoteur d’un certain transhumanisme regrette son geste : « Si je devais conseiller un jeune Craig Venter », je dirais, réfléchissez bien avant que vous veniez déverser votre génome sur Internet« …
Quelques questions centrales demeurent et l’une d’elle consiste à envisager que le génome d’une personne n’est pas du ressort de sa seule propriété… en effet, rendre disponible son génome revient à rendre disponible une partie des informations de ces enfants et des enfants de ceux-ci etc. Effectivement, les promoteurs de la génomique à large échelle envisagent de dépasser les problématique de l’héritabilité cachée (à ce sujet, lire l’excellent article de Bertrand Jordan dans M/S : Le déclin de l’empire des GWAS). Voici un extrait très pertinent qui explicite ce problème : « Les identifications réalisées dans le cadre des études GWAS sont certes scientifiquement valables et utiles pour la compréhension du mécanisme pathogène (donc porteuses d’espoirs thérapeutiques), mais, rendant compte de moins d’un dixième des héritabilités constatées, elles passent visiblement à côté d’un phénomène important… Comment résoudre ce paradoxe ? Il faut pour cela revenir sur ce qu’examinent réellement les GWAS. Elles se limitent aux Snip, faisant (pour le moment du moins) l’impasse sur les copy number variations (CNV), ces délétions, duplications ou inversions dont on a découvert récemment plusieurs centaines de milliers dans notre génome. Et même pour les Snip, elles ne donnent pas une image complète des variations génétiques entre individus. Par la force des choses, les 500 000 Snip représentés sur les puces d’Affymetrix ou d’Illumina (et qui ont préalablement été étudiés par le consortium HapMap) correspondent à des poymorphismes assez facilement repérables dans un échantillon de population : la règle adoptée a été de ne retenir que les Snip pour lesquels la fréquence de l’allèle mineur est au moins égale à 5 %. Cet usage était nécessaire pour limiter les difficultés dans le positionnement des Snip lors de l’établissement des cartes d’haplotypes ; mais il a pour conséquences que les GWAS n’examinent que les variants fréquents… Selon une hypothèse largement répandue, les maladies multigéniques fréquentes (diabète, hypertension, schizophrénie…) seraient dues à la conjonction de plusieurs allèles eux aussi fréquents : c’est la règle « common disease, common variant » souvent évoquée depuis une dizaine d’années. Les résultats de la centaine d’études d’association pangénomiques pratiquées à ce jour indiquent que cette hypothèse est très probablement fausse : les variants communs ne rendant compte que d’une faible partie de l’héritabilité, le reste est vraisemblablement dû à des variants rares (ponctuels ou non) dont ces études ne tiennent pas compte puisque les puces utilisées ne les voient pas. »
Ainsi, pour franchir ce cap, une solution simple est envisagée : le changement de résolution avec pour credo le passage de profils génomiques (quelques millions de SNPs) à l’intégralité du génome… et après l’épigénome et en même temps le métagénome. Si ces sciences bâties sur une technologie en pleine révolution permettent l’accès à un patrimoine humain universel (l’information génomique quasi exhaustive), si ces sciences renouvellent sans cesse leurs promesses -il faut des fonds et donc convaincre les pouvoirs publics pour acheter la technologie américaine qui permet d’accomplir ces sciences- hypothéquer le patrimoine humain ou pire le privatiser pourrait être une erreur dramatique dont on a du mal à mesurer l’étendue des conséquences.
![]() Une jeune start-up est née en mai 2012 : cette société issue du CNRS est porteuse d’une innovation dans le secteur du séquençage haut-débit. Enfin, une alternative française aux anglosaxons qui sont présents sur le marché depuis une dizaine d’année ! Maintenant… espérons que ce nouveau né n’arrive pas trop tard sur un marché animé par des fournisseurs de séquenceurs de 2ème génération (un marché mature) et d’autres fournissant des solutions de 3ème génération, riches de promesses.
Une jeune start-up est née en mai 2012 : cette société issue du CNRS est porteuse d’une innovation dans le secteur du séquençage haut-débit. Enfin, une alternative française aux anglosaxons qui sont présents sur le marché depuis une dizaine d’année ! Maintenant… espérons que ce nouveau né n’arrive pas trop tard sur un marché animé par des fournisseurs de séquenceurs de 2ème génération (un marché mature) et d’autres fournissant des solutions de 3ème génération, riches de promesses.
PicoSeq derrière ce nom emprunt d’humilité se cache une technologie de séquençage des plus ingénieuses : en effet, SIMDEQ™ (SIngle-molecule Magnetic DEtection and Quantification) la technologie de PicoSeq utilise une approche biophysique pour extraire des informations à partir de la séquence d’ADN ou d’ARN.
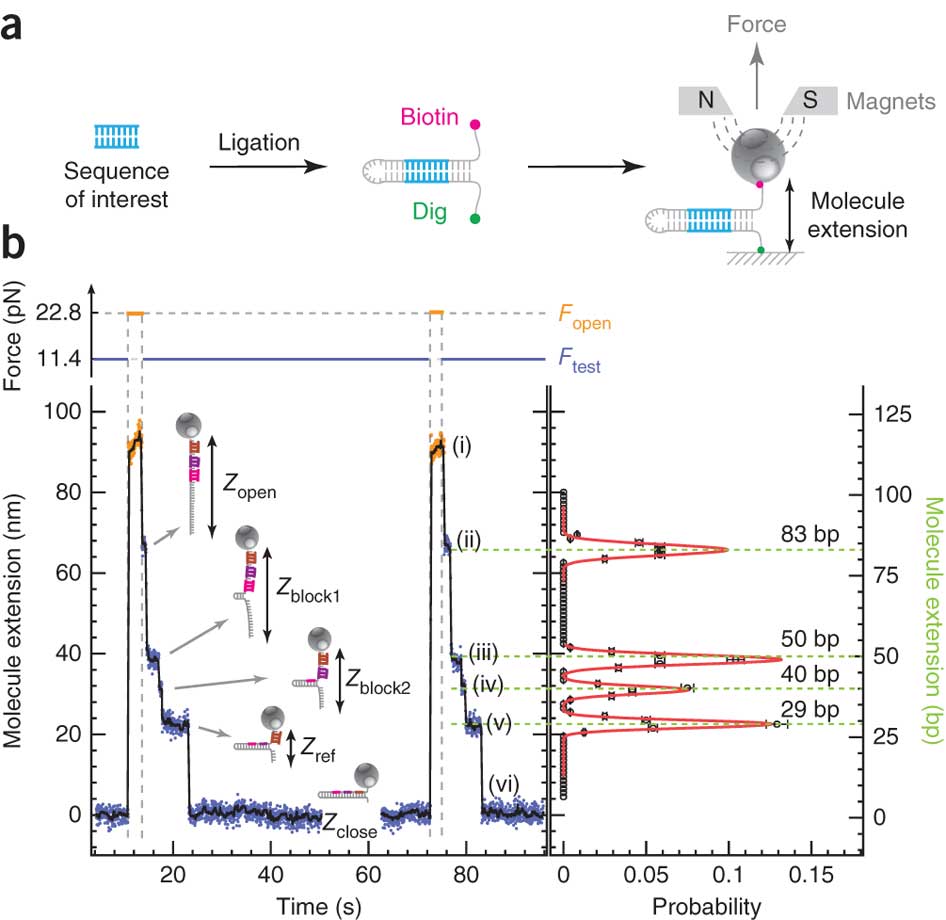
En s’appuyant sur cette représentation schématique tirée de Ding et al. (Nature Methods, 2012), on y voit un peu plus clair. Des fragments d’ADN ou d’ARN que l’on souhaite analyser servent de matrice pour la réalisation d’une librairie en «épingle à cheveux». Pour chaque épingle à cheveux, un côté d’un brin d’acide nucléique est attaché sur une surface solide plane et l’autre à une bille magnétique. En plaçant les billes dans un champ magnétique, modulant celui-ci de manière cyclique, les épingles à cheveux peuvent être auto-hybridées ou non (zip ou unzip). Ce processus peut être effectué des milliers de fois sans endommager les molécules constitutives de la librairie. La position de chaque bille est suivie à très haute précision permettant de voir ce processus d’ouverture et de fermeture en temps réel: nous avons donc là un signal brut permettant, en fonction de la force appliquée pour ouvrir totalement l’épingle à cheveu et des séquences d’oligonucléotides séquentiellement introduites dans le système de modifier la distance bille-support et de jouer sur le temps nécessaire où la force s’applique pour ouvrir l’épingle à cheveu…
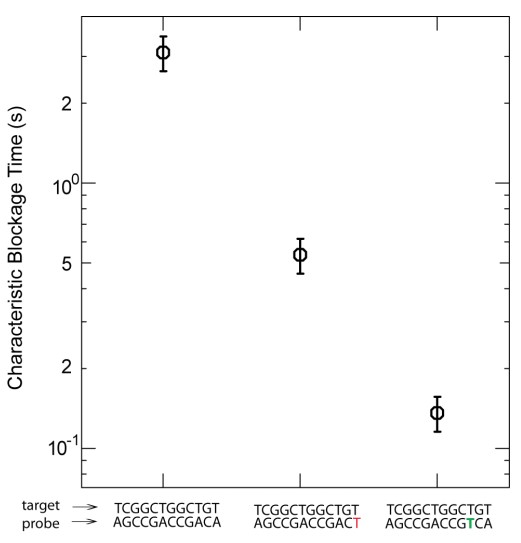
La figure ci-dessus (présente dans les données supplémentaires de l’article sus-cité) permet d’appréhender le potentiel de discrimination de la méthode… où le temps de blocage est fonction du nombre de mésappariements et de la position de ces mésappariements…
Finalement si l’aspect technique est intéressant puisqu’en rupture avec les méthodes proposées par PacBio certainement un peu moins avec le système proposé par Oxford Nanopore Technologies, si la perspective annoncée par PicoSeq est réellement séduisante: l’accès modifications épigénétiques de l’ADN, la question centrale est de savoir si le pas de la commercialisation (dans des conditions propices au succès) d’un tel outil, sera franchi.
Un article d’ Atlantico de septembre 2015, titré : les trois raisons pour lesquelles la France est incapable de rivaliser avec les géants américains de l’analyse ADN, est assez éclairant pour imaginer comment la concrétisation d’une preuve de concept peut être un chemin ubuescokafkaïen. Pour illustrer cela les propos de Gordon Hamilton, le directeur de la startup PicoSeq qui s’inquiète sur les entraves « typically french » peuvent faire office de témoignage. Ce dernier s’inquiète : « La qualité de la recherche scientifique (en France) est incroyable, l’une des meilleures » « le seul souci, c’est que l’on a beaucoup de difficultés ici à transformer ces recherches en vrai business » pour finir par citer en exemple les lenteurs administratives spécificités latines : « nous avons mis presque deux ans pour négocier les licences nécessaires aux brevets de Picoseq. Le même processus en Californie prend entre deux semaines et deux mois. Sur un marché aussi rapide que celui-ci, deux ans c’est très long. Tout change vite, c’est donc impossible pour nous d’être de sérieux concurrents de ces sociétés américaines qui ont toujours un temps d’avance ».
 Oxford Nanopore Technologies (ONT) crée un nouveau marché pour le séquençage haut-débit: le séquençage (haut-débit ?) à la portée de tous et en mode tout-terrain… mais pourquoi faire ? Le principe de cette technologie a été abordé dans plusieurs de nos articles : les données produites sont constituées de longs reads (quelques milliers de bases frôlant les 10 kB de moyenne), des reads assez bruités au-delà de 10 % d’erreurs; suffisamment longs pour permettre une identification quasi-certaine mais encore trop bruité (et trop peu profond dans le format portable MinION et maintenant SmidgION) pour pratiquer un bel assemblage de novo.
Oxford Nanopore Technologies (ONT) crée un nouveau marché pour le séquençage haut-débit: le séquençage (haut-débit ?) à la portée de tous et en mode tout-terrain… mais pourquoi faire ? Le principe de cette technologie a été abordé dans plusieurs de nos articles : les données produites sont constituées de longs reads (quelques milliers de bases frôlant les 10 kB de moyenne), des reads assez bruités au-delà de 10 % d’erreurs; suffisamment longs pour permettre une identification quasi-certaine mais encore trop bruité (et trop peu profond dans le format portable MinION et maintenant SmidgION) pour pratiquer un bel assemblage de novo.
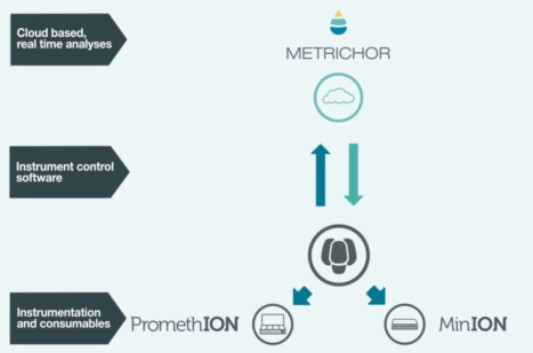
Donc imaginez vous, perdu au fin fond de l’Amazonie à la recherche de cette plante évoquée par le « sorcier » de la tribu Mashco-Piro que vous venez de quitter, plante potentiellement inconnue de notre médecine occidentale… Qu’à cela ne tienne! vous marchez en quête de la dite plante, vous, votre panneau solaire, votre smartphone, vos appareils ONT (Voltrax + SmidgION). Quasi certain de vérifier in situ votre trouvaille à l’aide d’un séquençage de 3ème génération. Enfin, ceci serait parfait si l’on oublie que Metrichor (le BaseCaller d’ONT) fonctionne en ligne… (cf schéma ci-dessous)… malgré les rêves les plus fous de Google et autres Facebook le fin fond de l’Amazonie n’est pas couvert par la 4G ! On peut imaginer qu’une application pour nos téléphones intelligents devra accompagner la mise sur le marché de cette suite d’appareils pour permettre une analyse en mode stand alone.
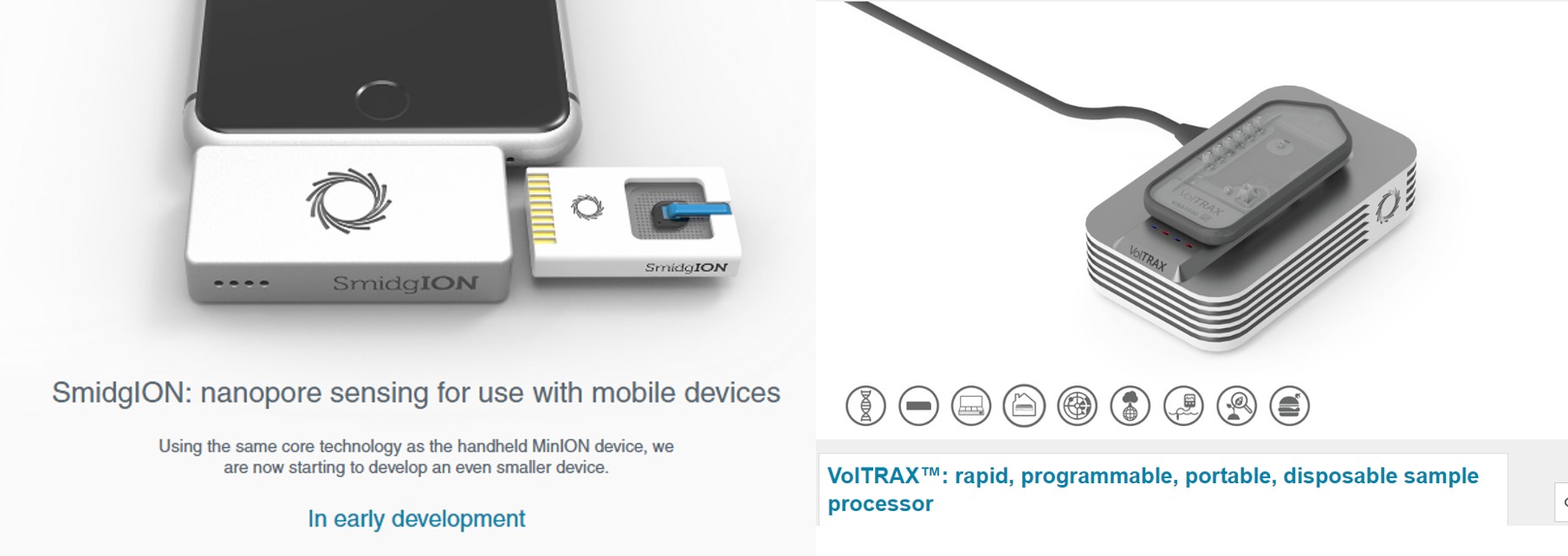
VolTRAX, l’une des promesses d’ONT, permet d’envisager la préparation de librairies à séquencer, ceci même perdu en pleine brousse. Par exemple, les nouveaux kits développés permettent une préparation d’une librairie en une dizaine de minutes. Que vous deviez séquencer un isolat du virus Zika ou Ebola sur le terrain (la logistique et le temps sont comptés) ou que vous deviez séquencer dans votre laboratoire favori, ce type d’automates permettant de simplifier les opérations relatives à l’élaboration de librairies de séquençage est souvent bien accueillis par les techniciens qui pourront s’adonner à des activités plus pertinentes.
Une heatmap (carte de densités) suite à un séquençage Ion Torrent évalue la densité de billes vivantes (porteuses d’une, ou malheureusement plusieurs, dans le cas de billes polyclonales, séquences matrice de séquençage).
Plus la surface de la puce de séquençage tire vers le rouge sombre plus important devrait être le nombre de reads séquencés. A l’inverse, le bleu sera indicateur d’une zone non pourvue de billes vivantes ou de billes tout court.
Ce premier indicateur de « qualité » de run est très précoce puisqu’il n’est pas nécessaire d’attendre la fin du run pour l’obtenir… ainsi une surface de puce en dégradés de bleus ne vous laissera aucune chance quant à l’exploitation des données (volume trop faible de reads). Il est à noter que la seule lecture de la séquence clé TCAG (portée par la matrice de séquençage) permet à la suite logiciel du PGM Ion Torrent de définir une bille vivante. Ainsi le rouge intégral est loin d’être un indicateur suffisant d’un séquençage satisfaisant.
Aujourd’hui à titre d’exemple nous avons obtenu lors de notre run cette heatmap (photo_Gaël Even) :

En cancérologie, l’allogreffe de moelle osseuse s’inscrit dans un parcours thérapeutique notamment comme traitement de consolidation après une chimiothérapie. Aussitôt, les notions de rejet ou d’acceptation du greffon apparaissent et il devient indispensable que les systèmes HLA (Human Leucocyt Antigens, découvert en 1950) du donneur et du receveur soient les plus proches possibles.
Ce système immunogène, situé sur le bras court du chromosome 6 chez l’homme, est caractérisé par son polygénisme et son polymorphisme qui sont à l’origine d’une grande variabilité interindividuelle et en fait le déterminant principal du résultat de greffe. L’ensemble des gènes HLA sont subdivisés en trois régions du chromosome 6 qui contiennent chacune de nombreux gènes avec ou sans fonction immunologique. On distingue ainsi la région CMH de classe I, de classe II, et de classe III.
A ce jour, un rendu de typage est ciblé sur une portion génomique restreinte codant pour le HLA. Il s’agit de l’exon 2 et 3 des loci HLA-A, HLA-B et HLA-C (région I), l’exon 2 et 3 des loci HLA-DQ (DQ-A et DQ-B) et l’exon 2 pour HLA-DR (DRA et DRB1), où repose prés de 70% du polymorphisme. La région III ne renfermant pas de gènes intervenant dans la présentation antigénique.

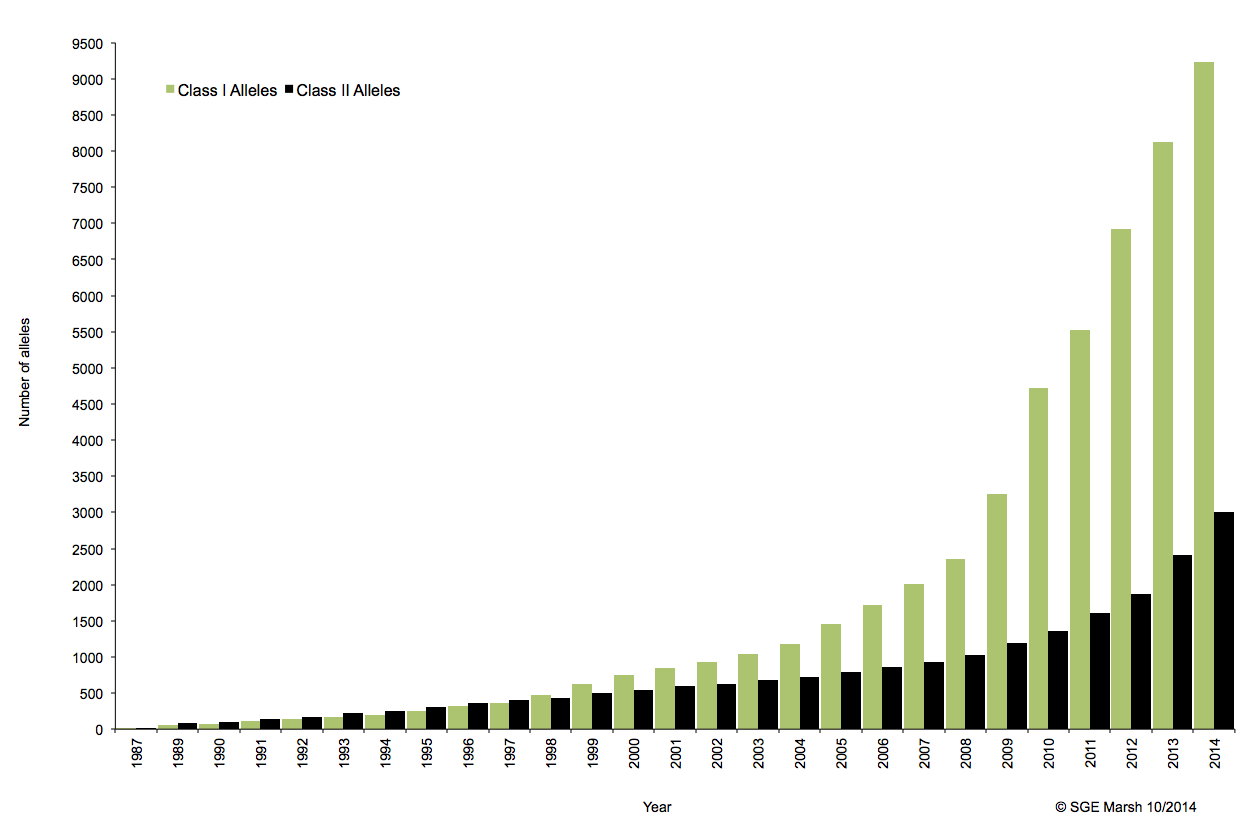
C’est ainsi que différentes approches de typages ont été développées et l’avènement de la technique de PCR au milieu des années 1980 a pallié aux limites de résolution de la sérologie employée jusqu’alors. D’un typage rendu au niveau générique (2 Digits), la « PCR-SSO » (Sequence Specific Oligonucleotide) et la « PCR-SSP » (Sequence Specific Primer) développées dans les années 1990 ont permis d’accéder à un résultat allélique (4 Digits). Cette avancée technologique s’est poursuivie la décennie suivante avec la « PCR-SBT » (Sequence Based Typing) ou séquençage « Sanger » puis plus récemment avec la PCR en temps réel (ex: linkage). Toutes ces techniques de biologie moléculaire ont permis de mettre en avant le polymorphisme et la grande diversité génétique du HLA. Chaque année, de nombreux allèles sont découverts, alimentant continuellement la banque de données de référence IMGT.

Associé à cette augmentation constante du nombre d’allèles typés, le nombre d’ambigüités croît et met progressivement en difficulté les technologies conventionnelles qui atteignent leurs limites. De plus, le pourcentage de réussite des allogreffes n’atteint environ que 50%. L’exploitation du reste de l’information génomique permettrait potentiellement d’améliorer cette performance par un typage plus affiné.
Le recours au séquençage nouvelle génération apparait donc inévitable. En plus de gérer les ambiguïtés par un séquençage allélique, le NGS permet de traiter simultanément de grandes quantités d’échantillons réduisant ainsi le coût unitaire, d’accéder à un niveau de résolution supérieur (4,6 ou 8 digits) tout en ayant la capacité de cibler des loci entier (Long Range PCR).
Ainsi, plusieurs stratégies existent avec leur solution technique adaptée à la préparation de la matrice d’ADN à séquencer:
– L’amplification ciblée des régions d’intérêt par PCR de fusion. Cette approche permets un gain de temps et une réduction des coûts en s’affranchissant des étapes de fragmentation, ligation, et autres purifications… .Par ailleurs elle n’est pas la mieux adapté dans le cas d’une couverture de séquençage de l’ensemble de la région génomique HLA.
– L’amplification par « Long Range PCR » permet une couverture complète des différents loci étudiés. Les fragments de plusieurs Kb subissent alors une fragmentation, une ligation des adaptateurs et indexation. Cette approche permet d’accéder à davantage d’informations (régions exoniques et introniques).
– La capture de séquences par hybridation. Même si cette solution est bien caractérisée elle n’est pas si efficace en terme de capture avec une disparité selon la taille des fragments.
– Le séquençage de génome entier ou d’exome. Cette approche est la moins biaisée et couvre tous les gènes du système HLA. Paradoxalement, l’analyse nécessite beaucoup trop de ressources pour une utilisation en routine et ne permets pas de traiter autant d’échantillons simultanément.
Plusieurs sociétés commerciales proposent des solutions clé en main depuis la préparation des échantillons (avec l’option « Long Range PCR » qui semble la plus plébiscité) jusqu’à l’analyse de résultats via leur logiciel dédié. Certaines sont en cours de validation de méthode pendant que d’autres tentent d’inonder le marché. Parmi elles, Gendx, Omixon, Illumina, One lambda, Life technologies, Immucor, etc…

L’exploitation des capacités et caractéristiques des solutions de séquençage à haut-débit permettrait d’affiner considérablement le typage HLA. Ainsi le décryptage de l’ensemble des régions codantes et non-codantes du génome d’intérêt représente un enjeu important dans la réussite des greffes. Par ailleurs, cette approche nécessitera une mise à jour considérable des banques de données (IMGT) avec une validation de nombreux nouveaux allèles.
Au cours des dix dernières années, la génomique connait une avancée technologique indéniable au travers des différents procédés de séquençage à haut-débit de deuxième génération. Néanmoins, certaines limites techniques subsistent, notamment par rapport à la quantité d’ADN requit, impliquant donc son extraction à partir de plusieurs millions de cellules. Cette contrainte implique une dilution de l’information pour des cellules aux fonctions biologiques bien souvent hétérogènes jusqu’au sein d’un même tissu.
En attendant la démocratisation du séquençage de troisième génération (Séquençage ADN sans amplification clonale) aux caractéristiques techniques qui permettraient un virage vers la génomique à l’échelle de la cellule unique , des méthodes alternatives appliquées à la seconde génération se développent afin d’accéder à cette hétérogénéité cellulaire. Ces solutions s’accompagnent donc, en amont du séquençage, d’une inévitable amplification de l’ADN de la cellule, préalablement isolée soit microdissection laser, système de microfluidique (ex: C1 Fluidigm), cytométrie en flux, ou encore micropipettage.
Ce poste est donc l’occasion de présenter la méthode d’amplification MALBAC, pour Multiple Annealing and Looping Based Amplification Cycles (Science, Zong et al.).
La méthode MALBAC repose sur l’utilisation de primers spécifiques (séquence de 8 nucléotides variables s’hybridant aléatoirement sur l’échantillon, couplée à une séquence connue de 26 nucléotides) générant des amplicons aux extrémités complémentaires. Cette particularité favorise la formation d’une boucle, évitant ainsi aux brins néo-synthétisés de servir à nouveau de matrice à la PCR et d’engendrer un biais d’ amplification (contrairement à la MDA). Ces étapes d’amplifications quasi-linéaires sont répétées cinq fois, puis les amplicons sont amplifiés par PCR exponentielle classique, en amont du séquençage.

MALBAC favorise une meilleure couverture de séquençage ainsi qu’une amplification plus uniforme (cf représentation ci-dessous) . De par ses performances, elle surclasse les méthodes conventionnelles tel que PEP-PCR, DOP-PCR ou encore MDA, pour Multiple Displacement Amplification, méthode la plus répandue depuis dix ans. Jusqu’à 83% de couverture à 10X de profondeur contre 45% pour la MDA. Parmi les autres atouts, la quantité d’ADN initiale requise n’est que de 0.5pg (contre 1000pg pour la MDA) et la polymérase Bst associée connait un taux d’erreur de 1/10000 bases.
Les performances de cette méthode permettent ainsi de reconsidérer les études génomiques ciblant un matériel biologique rare. Parmi elles, l’analyse de cellules tumorales circulantes, de tissus microdisséqués, de cellules embryonnaires, de micro-organismes, de cellules foetales circulantes, , etc…
 Parmi les technologies dédiées à la génomique, l’ « Optical Mapping » fait figure d’outil qualifiable d’alternatif. Cette approche repose sur une représentation graphique des sites de restrictions enzymatiques au travers d’un génome complet.
Parmi les technologies dédiées à la génomique, l’ « Optical Mapping » fait figure d’outil qualifiable d’alternatif. Cette approche repose sur une représentation graphique des sites de restrictions enzymatiques au travers d’un génome complet.
Les applications concernent aussi bien la génomique comparative (détection des délétions, insertions, inversions ou translocations), que le typage de souches (comparaison des cartes de restrictions). Aussi, conjuguée aux technologies de séquençage à haut-débit, elle permet également de répondre aux illusions fréquentes de l’obtention d’un « draft » de génome d’intérêt, nouvellement séquencé. Actuellement, OpGEN est la seule société proposant une solution semi-automatisée de cette technologie.
Techniquement, l’ « Optical mapping » consiste en (Cf fig ci-dessous):
– Une immobilisation des fragments d’ADN génomique extraits (1) au sein de canaux intégrés dans un support dédié (Argus System – OpGen) (2).
– Chaque molécule subit une digestion enzymatique (endonucléase de restriction) générant des sites de clivage, symbolisés ci dessous par les espaces (3).
– Le logiciel d’analyse (MapSolver) convertit ces données optiques en cartes moléculaires unitaires (4), qui alignées, fournissent une carte de restriction consensus du génome étudié (5).
L’utilisation de cette méthode, dans la perspective d’un assemblage efficace de génome, ne cesse de croître. En effet, elle permet de pallier les limites des NGS (Homopolymères, zones de génome peu ou non couvert) qui ne permettent bien souvent d’aboutir qu’à un nombre restreint de contigs (3′).
Il convient alors de créer une carte de restriction, in silico, de ces contigs (4′), à leur tour alignés sur l’ « optical map » du génome, sur la base des sites de clivage. Cette comparaison permet alors de positionner les contigs entre eux, de les orienter et de mettre en lumière les hypothétiques gaps. Le scaffold des contigs ainsi établi, associé à un séquençage Sanger des gaps permettent ainsi d’aboutir à un « draft » du génome étudié.

L’ « optical mapping » apparait comme un outil fiable et utile dans l’assemblage de génome, d’autant qu’il fait appel à une technique différente, indépendante mais à la fois très complémentaire au séquençage à haut débit.
 A l’aube des années 2000, la génomique appliquée au domaine végétal fait l’objet d’une mobilisation internationale de grande ampleur comme en témoignent les programmes « Zygia » et « Gabi » en Allemagne, « Plant Genome Initiative » aux États-Unis, ou encore « Rice Genome Research Program » au Japon, qui poursuivent des objectifs analogues. Il devient primordial d’identifier les gènes et leur fonction jouant notamment un rôle décisif dans la production végétal (Rusticité, résistance aux maladies, aux herbicides, etc…).
A l’aube des années 2000, la génomique appliquée au domaine végétal fait l’objet d’une mobilisation internationale de grande ampleur comme en témoignent les programmes « Zygia » et « Gabi » en Allemagne, « Plant Genome Initiative » aux États-Unis, ou encore « Rice Genome Research Program » au Japon, qui poursuivent des objectifs analogues. Il devient primordial d’identifier les gènes et leur fonction jouant notamment un rôle décisif dans la production végétal (Rusticité, résistance aux maladies, aux herbicides, etc…).
Cette période est également marquée par l’achèvement du séquençage du génome de la plante modèle Arabidopsis thaliana, étape majeure dans la recherche en biologie végétale.
Simultanément, des collections de mutants d’insertions (T-DNA) chez A. thaliana sont créés au sein de nombreux groupes (SALK, GABI-Kat, Syngenta, INRA Versailles, etc…), et elles émergent notamment au travers du projet « Genoplante« , programme fédérateur en génomique végétale (Groupement d’Intérêt Scientifique regroupant à la fois des organismes publics tel que l’INRA, CNRS, Cirad, IRD et de puissants partenaires privés tel que Biogemma, Rhône-Poulenc Santé végétale et animale et Bioplante). L’idée est donc d’utiliser ces banques de mutants comme outils pour la génomique fonctionnelle appliquée à la plante modèle.
A l’époque, les solutions proposées pour l’identification des positions d’insertion du T-DNA au sein du génome sont nombreuses ( « Tail-PCR », « Inverse PCR », « Kanamycin Rescue » ). Néanmoins, ces approches restent fastidieuses: En plus de présenter certaines étapes techniques limitantes, elles sont également très chronophages.
Récemment, de nombreuses études ont commencé a démontrer l’énorme potentiel du séquençage à haut-débit dans l’identification des sites d’insertion de transposons. Le terme générique « Tn-Seq », pour « Transposon-Sequencing », est une variante du séquençage d’amplicons ciblés (Target-seq) et peut se décliner selon quatre méthodes comme illustrées ci-dessous (Tim van Opijnen and Andrew Camilli, Nature reviews – Microbiology (2013 July)). Elles dépendent notamment de la procédure de préparation de librairie de séquençage employée:
![]()
– Le »Tn-seq » et « INSeq » (respectivement pour « Transposon sequencing » et « Insertion sequencing ») sont deux approches très similaires reposant sur un séquençage d’amplicons obtenus à partir d’un couple d’oligos dont l’un cible le transposon. Seule la méthode de purification varie (Gel PAGE pour « INSeq » et Gel agarose pour « Tn-Seq)
– Le « HITS » et « TraDIS » (respectivement pour « High-throughput insertion tracking by deep sequencing » et « Transposon-directed insertion site sequencing ») sont également deux méthodes très similaires notamment en amont de l’étape de PCR de librairie.
L’alignement des données de séquençage (.fastq) sur le génome de référence, permet ainsi d’identifier la position du site d’insertion. L’illustration met en évidence les « reads » issus de la PCR de librairie ciblant les régions flanquantes au Transposon (« En vert » la bordure gauche, « en rouge » la bordure droite). Sur la base de cette méthode, il devient donc aisé d’identifier le nombre d’insertion potentielle.

L’utilisation des technologies de séquençage à haut-débit pour l’identification des sites d’insertion de T-DNA dans les banques de mutants révolutionnent les méthodes de criblage. Tout en s’affranchissant de techniques fastidieuses, cette approche de Tn-seq présente à la fois l’avantage de pouvoir gérer simultanément un très grand nombre d’échantillons (barcoding), à des coûts réduits et dans un délai des plus respectables.

En ce début d’année, cet article est l’occasion d’aborder rapidement les divers axes de développements, les différents acteurs du séquençage haut-débit de deuxième génération.
– Commençons par Life Technologies et sa gamme Ion Torrent. En fin d’année 2013, la Ion Community (forum où se retrouvent les utilisateurs de la technologie Ion Torrent) s’agite à l’annonce de 3 nouveautés majeures (early access program) :
(i) L’accès à une nouvelle chimie de séquençage, la Hi-Q ™, permettant d’accroître la fiabilité de séquençage. Les erreurs seraient réduites de 90 %, ceci même au niveau des homopolymères, et pour des reads de 400 bases, témoignage de Dag Harmsen à l’appui ! En clair, il semble que ce soit l’enzyme (what else ?) qui ait été remplacé.
(ii) La deuxième annonce concerne l’arrivée de la chimie Avalanche où plusieurs heures d’amplification clonale à l’aide d’un automate One-Touch peuvent être remplacées par l’emploi d’un tube, ce qui prend alors 2 heures pour obtenir une librairie de 500 pb, et ce, de façon isothermique. Un choc de simplification qui ravira les utilisateurs pour lesquels cette étape est limitante.
(iii) La troisième annonce concerne la mise à disposition de kits permettant de réaliser des analyses métagénomiques ciblées 16 S. Un système exploitant le PGM et sa capacité de produire des reads de 400 pb. L’inconnu ici réside dans la mise à disposition de la communauté d’un pipeline analytique performant.
– Qiagen, qui n’est pas connu pour être un acteur de poids sur la scène du séquençage haut-débit, arrive en force en cette année 2014 avec une solution intégrant tous les jalons nécessaires à la complétude d’une étude. Fort de son rachat d’une solution de séquençage (lire l’article : Qiagen investit… le séquençage haut-débit de 2ème génération), Qiagen propose un environnement logiciel des plus intéressants ! En effet, la société néerlandaise a racheté les sociétés CLC Bio et Ingenuity systems. Ces deux sociétés proposent l’une des toutes meilleures solutions d’analyse de séquences: une solution d’assemblage de novo réellement performante grâce à CLC genomics workbench, et Ingenuity systems proposant les pipelines d’analyses suivants: IPA, pour donner un sens biologique aux données omiques, Ireport pour l’analyse de données d’expression et Variant Analysis, un pipeline permettant d’optimiser la recherche de mutations causales.
Ainsi QIAGEN, à l’instar de ce que nous avons tâché de représenter par le schéma ci-dessous, possède actuellement tous les maillons (ou pas loin) d’une chaîne allant de l’échantillon à l’analyse finale traduisant des données de séquences en sens biologique.

– Illumina, quant à elle, semble avoir l’ambition de devenir une sorte de Apple de la « génomicosphère ». En effet, Illumina propose BaseSpace, un Itunes pour les biologistes. D’ailleurs, notons qu’Illumina propose sur Itunes une application : MyGenome, qui propose « d’explorer un véritable génome humain » et d’afficher des rapports sur les variations génétiques importantes. « L’application MyGenome fournit une interface simple, intuitive, et éducative pour vous lancer à la découverte du génome humain« . Revenons à BaseSpace, une interface entre vous et un cloud hébergeant des applications et des données permettant d’analyser les séquences en sortie de MiSeq ou HiSeq. Ce cloud permet aux utilisateurs de délocaliser le stockage de leurs données. L’idée : simplifier au maximum l’analyse par la mise à disposition d’outils et la mise en réseau des utilisateurs. Illumina s’est aperçu que si le goulot d’étranglement constitué par l’analyse de données de séquençage haut-débit volait en éclat, nécessairement les runs pourraient se multiplier avec leur chiffre d’affaire. Le schéma ci-dessous reprend quelques éléments de la solution analytique développée par Illumina.

Une communauté de plus de 12000 utilisateurs, un espace permettant l’utilisation d’une vingtaine d’applications. L’objectif d’Illumina : créer un espace attractif, émulant et incitant les intervenants à mettre à disposition les applications développées en priorité sur cet espace. Anticipant une demande exponentielle d’analyses et d’espace de stockage lorsque le HiSeq a été intégré au BaseSpace, Illumina a décidé de mettre en place une politique de tarification qui limiterait la quantité d’espace libre pour stocker et traiter les données génomiques dans le cloud. En vertu de cette logique, les utilisateurs reçoivent un téraoctet gratuit d’espace pour le stockage et le traitement des données et seraient alors en mesure d’acheter du stockage supplémentaire par incréments de téraoctet ou 10 téraoctets – un téraoctet coûterait 250 $ par mois ou $ 2,000 d’avance pour une année complète , tandis que 10 téraoctets seraient à 1500 $ par mois ou une avance des frais annuels de $ 12 000 (données chiffrées début 2013).
En conclusion, si les années précédentes ont vu le lancement de nouveaux séquenceurs, avec depuis 2011 l’arrivée de séquenceurs de paillasse, les années 2013-2014 attendent la diffusion de séquenceurs de 3ème génération. Qiagen est un petit nouveau dans la course, ce nouvel acteur est capable, sans réel développement, de proposer une solution complète grâce à une stratégie de rachat pertinente. Illumina et Life Technologies, pendant ce temps, poursuivent leur développement en essayant d’émuler les utilisateurs avec, respectivement, leur BaseSpace et Ion Community. L’opérateur historique, Roche est le grand silencieux avec une stratégie peu lisible…
 Le séquençage du génome humain pour 30$, c’est la promesse faite par David Weitz, co-fondateur de GnuBio au cours de l’année 2010. Trois ans plus tard, la start-up vient de lancer en béta-test son nouveau procédé de séquençage à haut débit. Il s’agit du premier système entièrement intégré (amplification des cibles, enrichissement, séquençage et analyse) qui propose pour le moment une application de target-sequencing destinée aux cliniciens et dédiée au diagnostic moléculaire.
Le séquençage du génome humain pour 30$, c’est la promesse faite par David Weitz, co-fondateur de GnuBio au cours de l’année 2010. Trois ans plus tard, la start-up vient de lancer en béta-test son nouveau procédé de séquençage à haut débit. Il s’agit du premier système entièrement intégré (amplification des cibles, enrichissement, séquençage et analyse) qui propose pour le moment une application de target-sequencing destinée aux cliniciens et dédiée au diagnostic moléculaire.
En 2010, David Weitz et son équipe de l’Université d’Harvard ambitionnent de développer une nouvelle technologie de séquençage à haut-débit, alliant les technologies de biologie moléculaire aux procédés de microfluidique développés quelques années plus tôt (2004) au sein de la société RainDance technologies.
Cette nouvelle approche repose sur la capacité à générer des gouttes de l’ordre du picolitre et pouvant être déplacée sur une puce microfluidique. Ces gouttes peuvent renfermer soit un couple d’amorces, des adaptateurs, ou tout autre type de réactifs nécessaires aux étapes de préparation de librairie et de séquençage (séquençage par hybridation-ligation, type SOLiD avec une fidélité de 99.99%). Dès lors, leurs quantités utilisées au sein de ces picogouttes sont considérablement revues à la baisse, ce qui constitue le point clé à une réduction des coûts de séquençage et donc la perspective d’un séquençage de génome humain à 30$, selon David Weitz.
Les projets de GnuBio sont désormais d’élargir le champs d’applications de leur séquenceur à l’analyse transcriptomique (RNA-seq), l’étude de la méthylation (ChiP-seq) ou encore le séquençage de génome entier. La société ambitionne une commercialisation de leur équipement au cours de l’année 2014.
A suivre…
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.


{kind=link}
{kind=link}
{kind=link}