
Currently viewing the tag: "Séquençage"

L’un des pionniers des séquences nucléique et protéique est décédé, il y a peu, le 19 novembre 2013, à l’âge de 95 ans. Il avait commencé sa thèse en octobre 1940 avec pour projet de rechercher une protéine consommable purifiée d’herbe (relativement abondante dans le royaume de Georges VI). C’est, au final, sous la direction d’Albert Neuberger (qui avait fuit l’Allemagne avec l’arrivée d’Hitler au pouvoir) que Frederick Sanger devient docteur en 1943.
Les deux hommes se respectaient, à propos de « son maître » Albert, Frédérick Sanger avait déclaré : « c’est Neuberger, qui le premier, m’a appris à faire de la recherche, à la fois techniquement et comme un mode de vie, et je lui dois beaucoup. »
L’homme qui a donné son nom à une technique de séquençage des nucléotides ouvrant la voie à un pan entier d’une science, à un institut de recherche (le Wellcome Trust Sanger Institute) employant plus de 900 personnes est une fierté (jamais anoblie) de l’empire britannique : une des quatre personnes à avoir reçu deux prix Nobel (prix Nobel de chimie). Son premier prix Nobel de chimie pour le séquençage de l’insuline (1958) sanctionne un travail de 10 ans. Rejoignant Crick (le penseur), au laboratoire de biologie moléculaire du Conseil médical de la Recherche à Cambridge, Sanger (le fonceur) se met à développer une technique de séquençage de l’ADN. Ce travail lui apportera son second prix Nobel en 1980.
« Quand j’étais jeune, mon père me disait que les deux activités les plus intéressantes dans la vie sont la recherche de la vérité et de la beauté, et je crois qu’Alfred Nobel a dû ressentir la même chose quand il a donné ces prix pour la littérature et les sciences.«

Un document reprenant le nécessaire à savoir sur l’oeuvre de Frederick Sanger (réalisé par Richard Daniellou, Enseignant-chercheur, ENSCR) est disponible au format pdf.
L’image en tête de cet article est issue d’un portrait de Frederick Sanger réalisé par Paula A. MacArthur (huile sur toile : 1991, collection: National Portrait Gallery de Londres).
 Ce qui suit pourrait être tiré d’un roman d’anticipation technofuturiste où le héros s’empêtre dans un polar aux implications géopolitiques. Après la mise en application de la politique de l’enfant unique, politique lancée en 1979 par Deng Xiaoping qui a eu la fâcheuse conséquence d’induire un déséquilibre entre les sexes (116 garçons pour 100 filles, 2005), la Chine, forte de sa population de presque 1 milliard 400 millions de ressortissants, cherche à appliquer les lois de la sélection génomique sur sa propre population. Le Q.I., phénotype relativement héritable (héritabilité supérieure à 0,5) est le deuxième caractère, après le sexe, « choisi » par les autorités. Cette article développe les différents éléments qui ont permis à la Chine de devenir le premier pays transhumaniste.
Ce qui suit pourrait être tiré d’un roman d’anticipation technofuturiste où le héros s’empêtre dans un polar aux implications géopolitiques. Après la mise en application de la politique de l’enfant unique, politique lancée en 1979 par Deng Xiaoping qui a eu la fâcheuse conséquence d’induire un déséquilibre entre les sexes (116 garçons pour 100 filles, 2005), la Chine, forte de sa population de presque 1 milliard 400 millions de ressortissants, cherche à appliquer les lois de la sélection génomique sur sa propre population. Le Q.I., phénotype relativement héritable (héritabilité supérieure à 0,5) est le deuxième caractère, après le sexe, « choisi » par les autorités. Cette article développe les différents éléments qui ont permis à la Chine de devenir le premier pays transhumaniste.
Acte 1 : une politique de reconquête
La Chine possède des ports en Europe, une partie significative de la dette américaine, des milliers d’hectares agricoles en Afrique et un parc de bombes atomiques qui ne laisserait pas insensible le docteur Folamour. Malgré un budget de millions de dollars alloué à une armée aux ordres du parti unique, l’usine du monde ne semble pas avoir de volonté expansionniste impérialiste en vue de repousser ses frontières (si on omet le sujet du Tibet). Il semble que les ambitions de la Chine actuelle résident moins dans le fait de mettre à la disposition de l’occident sa main-d’œuvre bon marché que de devenir la première puissance mondiale matérielle et intellectuelle. c’est pourquoi le pays mise toujours sur sa vraie matière première : l’humain. Devenue une puissance spatiale avec ses Taikonautes, la Chine envoie dans les meilleures universités du monde, ses étudiants qui, de retour (pour ceux qui reviennent), accomplissent une recherche de tout premier plan. Dans le champ de la génomique et des biotechnologies, la Chine s’impose aujourd’hui comme un acteur de tout premier plan.
Acte 2 : une recherche efficace
En effet, la Chine compte plus d’un cinquième de la population mondiale et compte aussi sur la volonté de fer pour retrouver son lustre d’antan. Il est alors, relativement aisé d’imaginer que même si une portion faible de sa population atteint les études supérieures, le pays sélectionne des étudiants qui deviendront des chercheurs motivés et ayant émergé des tous meilleurs étudiants d’une classe d’âge donné. Il est passé le temps où les meilleurs chercheurs chinois s’exilaient inconditionnellement aux Etats-Unis pour finir par être naturalisés – le classement académique des universités mondiales (aussi appelé classement de Shanghaï, ou encore « Academic Ranking of World Universities » en anglais) est un outil parfait pour identifier les meilleures universités où envoyer ses étudiants. La Chine devient même attractive sur la scène mondiale de la recherche scientifique et technologique. Selon un rapport de l’OCDE, le pays est deuxième pour la dépense en recherche et développement, injectant 136 milliards US$ en 2006 (USA : 330 milliards US$ pour la même période).
L’Empire du milieu oriente sa politique du développement technologique dans des domaines jugés comme hautement stratégiques: l’énergie, les ressources naturelles, l’environnement, les techniques de production industrielle et les technologies de l’information, les biotechnologies, les nanotechnologies, les technologies spatiales et maritimes. La Chine est en tête des pays en nombre de brevets déposés par année, ceci est l’aboutissement de trente ans durant lesquels le pays a misé sur la recherche scientifique pour rattraper son retard technologique sur l’Occident. Depuis le début des années 2000, le gouvernement promeut une innovation purement chinoise. Concernant le niveau des étudiants chinois, ceux-ci commencent à être l’objet de discriminations dans certaines universités américaines où ils occupent les meilleurs places. La recherche scientifique, basée sur une « exportation » massive d’étudiants a permis à la Chine de rattraper son retard technologique, cette technologie aujourd’hui sert une recherche scientifique de tout premier plan et sert, en ce qui concerne la biotechnologie, un dessein qui peut faire peur, vu d’ici.
Acte 3 : des moyens puissants
L’exemple qui vient à l’esprit immédiatement est le BGI (Beijing Genomics Institute). A ce sujet, vous pouvez lire notre précédent article : Taylor + Deng Xiao Ping + Watson = la Chine marche sur les chromosomes. Rappelons que le BGI est la plus grande plateforme génomique mondiale et un contributeur scientifique de tout premier ordre. Grâce à des plateformes de haute-technologie permettant de générer des volumes de séquences et de typages à de très bas coût, la Chine aurait la possibilité d’appliquer les résultats de sa recherche scientifique à l’échelle de sa vaste population.

Le BGI développe un laboratoire de génétique cognitive avec pour ambition de répondre à ces questions : Comment fonctionne le cerveau humain ? Comment les gènes affectent la capacité cognitive ? Comment les gènes et l’environnement interagissent pour produire l’intelligence et la personnalité humaine ? Le BGI a lancé un recrutement d’une cohorte de personnes volontaires aux Q.I. élevés. L’objectif est donc de rechercher les variations génétiques associées à des variations significatives au niveau du Q.I. (notons qu’une étude cas-témoin eut nécessité une cohorte de Q.I. bas à très bas… difficile de procéder à un recrutement volontaire !). Le Q.I. étant plutôt héritable, il sera donc techniquement possible de sélectionner les porteurs des variants « Q.I. élevé ». Ainsi il est possible de procéder à deux niveaux :
– orientation / optimisation « d’accouplements » : les porteurs de ces variations « Q.I. élevé », après s’être génotypés, auront tendance à rechercher un conjoint lui aussi porteur de ces mêmes variations afin de maximiser leur chance de procréer un enfant lui même porteur de ces variations (notons ici qu’il n’y a pas de réel progrès génétique, en tant que tel)
– passons par l’étape FIV : évitant de miser sur le hasard, les gamètes des deux futurs parents produiront plusieurs dizaines d’embryons, chaque œuf sera biopsié (une petite étape de phi29, un génotypage ciblé ou une sélection génomique sur la base de l’exploitation d’un génotypage pan-génomique à l’aide d’une cartographie basée sur plusieurs millions de marqueurs SNPs humains). Fort de la connaissance du « Q.I. estimé » de ces dizaines d’œufs… il ne restera qu’à « contrecarrer » l’aléa de méiose et de ne ré-implanter que ceux présentant le meilleur score de Q.I. estimé…
![]() La sélection humaine pose bien évidemment des problème de bioéthique. Si l’occident sélectionne d’ores et déjà des êtres humains dans le but de limiter les enfants nés porteurs de tares génétiques majeures, l’Empire du milieu semble parti sur une autre voie (même si cette voie n’empêche pas l’autre) : améliorer significativement le Q.I. de sa population et ainsi basculer dans l’ère de la transhumanisation (selon ce mouvement intellectuel, l’Homme doit prendre en main sa propre évolution).
La sélection humaine pose bien évidemment des problème de bioéthique. Si l’occident sélectionne d’ores et déjà des êtres humains dans le but de limiter les enfants nés porteurs de tares génétiques majeures, l’Empire du milieu semble parti sur une autre voie (même si cette voie n’empêche pas l’autre) : améliorer significativement le Q.I. de sa population et ainsi basculer dans l’ère de la transhumanisation (selon ce mouvement intellectuel, l’Homme doit prendre en main sa propre évolution).
Après le Q.I., le grand jeu du moment consiste à deviner quel sera le phénotype de choix (la docilité ?, le courage ?), il faut espérer que l’on attendra quelques décennies avant d’envisager le premier humain OGM… Envisageant cela, on caricature la démarche actuelle chinoise et on s’imagine, un peu, à la place de Charlton Heston à la fin de Soleil Vert (film de Richard Fleischer, sorti en 1973) quand il découvre la matière première constitutive de l’aliment qui nourrit une planète devenue stérile.
 A l’heure où Jonathan Rohtberg et Craig Venter cherchent de l’ADN sur Mars (lire : Searching for Alien Genomes), l’ADN sur Terre ne serait-il pas à la veille de changer de statut ? En effet, si l’ADN est « le support de l’information génétique »… est-il possible que cette macromolécule devienne le support de l’information tout court ?
A l’heure où Jonathan Rohtberg et Craig Venter cherchent de l’ADN sur Mars (lire : Searching for Alien Genomes), l’ADN sur Terre ne serait-il pas à la veille de changer de statut ? En effet, si l’ADN est « le support de l’information génétique »… est-il possible que cette macromolécule devienne le support de l’information tout court ?
– C’est humblement, dans le domaine de la traçabilité, que l’ADN a trouvé un rôle d’espion : Ainsi, au début des années 2000, la société norvégienne ChemTag, tente de développer des systèmes permettant de tracer dans nos océans, le pétrole retrouvé suite à d’éventuels dégazages. L’idée est simple : l’ADN est utilisé comme un code-barre, la succession du code de quatre lettres permet assez rapidement d’obtenir un code (4^n) ne permettant qu’avec une probabilité infime d’être retrouvé par hasard. Il faut savoir que par année, plus d’un million de tonnes de pétrole sont déversées dans les océans. La problématique en vaut la peine, Poséïdon en sera gré. Ici toute la difficulté qui se présentait à la société ChemTag : faire en sorte que l’ADN ait une grande affinité pour le pétrole (évidemment en cas de dégazage si le traceur ADN se dissout dans l’océan… adieu code-barre) et permettre que ce traceur soit purifié le plus simplement possible. L’ADN est une molécule plutôt stable et plutôt facile à « lire », ne modifiant pas les qualités organoleptiques d’une substance… et surtout cette molécule permet un nombre infini de combinaisons.
Moult industriels souhaiteraient être capables d’identifier en toute objectivité (légalement) tout organisme vivant leur appartenant (une souche de production par exemple). A cette fin, le code barre biologique peut être utilisé de deux manières :
– artificiellement, en « incorporant » par manipulation génétique une séquence connue. Cette option est bien souvent écartée parce qu’elle fait appel à la notion d’OGM, mention qui peut effrayer le consommateur.
– naturellement, en connaissant le patrimoine génétique du génome employé dans un procédé industriel. Cela implique de déterminer une séquence singulière qui n’appartiendrait qu’à cette organisme. Souvent ce type de procédures fait appel à une combinaison de séquences ou de loci polymorphes.
L’évolution des techniques de séquençages (augmentation des débits, diminutions des coûts, rapidités des runs) a permis de rendre accessible une séquence complète d’un micro-organisme ou une carte de SNPs haute-densité pour certains eucaryotes supérieurs dont on dispose de puces SNPs.
Aujourd’hui, certains envisagent d’exploiter l’ADN pour en faire un support robuste de l’information numérique.

Depuis quelques semaines, une petite bataille a lieu entre l’équipe de Georges Church de la Medical School de Boston et Christophe Dessimoz de l’EBI.
Le premier a encodé un livre dont il est le co-auteur : Regenesis: How Synthetic Biology Will Reinvent Nature and Ourselves in DNA comportant 53000 mots et 11 images en jpeg accompagné d’un programme Javascript pour un total de 5,37 Mo dans un picogramme d’ADN (un peu moins de 1 Gbase). Le second a encodé du Shakespeare, un discours de Martin Luther King, une photo et la copie d’un article de 1953 décrivant la structure de l’ADN… on s’amuse plutôt pas mal aussi de ce côté de l’Atlantique !
Ces querelles d’encodeurs ont malgré tout un intérêt. Ils démontrent la faisabilité de ce type de système et montrent qu’il est désormais envisageable de sauvegarder pour des échelles de temps très longues, de grandes quantités d’informations qui échapperaient à bien des autodafés.
Les systèmes d’encodage au sein même de l’ADN relégueront nos séquenceurs haut-débit au niveau de lecteurs DVD ou Blu Ray du futur… La limite du système, actuellement, tient plus des synthétiseurs d’ADN et des technologies de séquençage qui n’ont pas réellement été conçus à cette fin. Mais à l’heure où l’on promet d’ici quelques dizaines de mois un séquençage de génome humain pour une centaine de dollars contre quelques milliards il y a 10 ans, tous les espoirs sont permis pour que l’information séculaire contenue dans une bibliothèque patrimoine de l’Humanité devienne une bibliothèque d’Alexandrie du futur, franchissant les âges, échappant aux catastrophes naturelles, aux censures et à l’oubli.
Le terme « Nouvelle génération de séquençage à haut-débit » ( ou « Next generation sequencing » ) regroupe l’ensemble des technologies ou plateformes de séquençage développées depuis 2005 par quelques sociétés de biotechnologies.
L’objectif de cet article est de proposer de manière synthétique, un tour d’horizon des différents principes et caractéristiques de ces nouveaux outils et ainsi fournir quelques orientations et solutions techniques en réponses à des questions biologiques.
La position actuelle dans laquelle nous nous trouvons, entre la commercialisation de certains séquenceurs et ceux en cours de développement, est caractéristique d’une période charnière dissociant les technologies à haut débit dites de 2ème génération qui requièrent une étape d’amplification des molécules d’ADN en amont du décodage, de celles dites de 3ème génération permettant le décryptage direct d’une seule molécule d’ADN. Cette dernière catégorie fera à elle seule l’objet d’un prochain article.
Le marché des séquenceurs de 2éme génération est couvert par 3 grand groupes que sont Roche, Illumina et Life Technologies ayant respectivement proposés de manière successive, leur première plateforme à savoir le 454, le Genome Analyser et enfin le SOLiD. Depuis, le marché s’est étoffé proposant un panel de technologies au principe et caractéristiques propres telles qu’elles sont mentionnées ci dessous. A noter que parmi ce panel, le PGM, Ion torrent est le seul a connaitre une évolution constante en terme de capacités de séquençage (10Mb – reads de 100b – Juin 2011 / 100Mb – reads de 100b – Sept 2011 / 100Mb – reads 200b – Nov 2011 / 1Gb – Jan 2012 )

Chaque plateforme possède ses avantages et inconvénients et nombreuses sont celles configurées pour répondre à de nombreuses approches « omics », dans certaines limites. Il s’agira de faire un choix technologique selon les champs d’applications souhaités.
De manière générale, le type d’organisme étudié prédéterminera la technologie à employer. La notion de profondeur est récurrente à chaque application et dans l’objectif d’un reséquençage, le choix de la plateforme peut être identifié, de manière simplifiée, sur la base d’un calcul rapide ( P = N / L où P: Profondeur, N: Nombre des nucléotides totaux des reads, L: Taille du génome étudié).
Concernant les séquenceurs de 2ème génération, le séquençage de novo est une application mentionnée chez de nombreux fournisseurs (cf le tableau ci-dessous). Toutefois, l’association de deux technologies générant à la fois des reads longs (type 454, Roche) et une profondeur conséquente (type GAIIx, Illumina) palliant aux problèmes liés aux homopolymères et erreurs de séquençage, est préconisée (Au cours de l’article à venir sur les séquenceurs de 3ème génération, nous aborderons les plateformes davantage configurées pour cette application).
Ce paramètre de profondeur sera également à prendre en considération pour les champs d’applications incluant la notion d’analyse quantitative (RNAseq, ChIPseq, …). Si la profondeur permet d’atténuer les erreurs de séquençage, il reste néanmoins préférable de s’orienter vers des technologies à Q30 minimum (1 erreur sur 1000) pour la détection de SNPs.

Selon les technologies évoquées ci-dessus, les caractéristiques et champs d’applications ont évolués. Aussi, je vous propose de retrouver l’ensemble de ces informations actualisées en cliquant sur ce lien.
L’ensemble des informations sont détaillées dans l’article mentionné ci-dessous:

Voici les résultats, présentés lors des journée France Grille 2011, d’une étude sur le déploiement de traitement de données NGS sur grille de calcul menée par plusieurs plateformes de bioinformatiques et centres de calcul universitaires français dans le cadre du projet GRISBI (Grille Support pour la Bio-Informatique) :
GRISBI a  pour but, à travers la mutualisation de ressources informatiques des acteurs du projet, de proposer l’utilisation d’applications bioinformatiques déployées sur grille de calcul au plus grand nombre.
pour but, à travers la mutualisation de ressources informatiques des acteurs du projet, de proposer l’utilisation d’applications bioinformatiques déployées sur grille de calcul au plus grand nombre.
pour but, à travers la mutualisation de ressources informatiques des acteurs du projet, de proposer l’utilisation d’applications bioinformatiques déployées sur grille de calcul au plus grand nombre.Le constat est simple : la gestion des données provenant des séquenceurs, autant dans le stockage que l’analyse, va de paire avec la mise en place de nouvelles infrastructures plus efficaces, plus adaptables, plus sécurisées, plus accessibles que des solutions locales : les grilles de calculs sont une réponse à beaucoup de ces problématiques.
On nous présente les premiers résultats obtenues suite à un assemblage denovo sur ABySS et Ray et un assemblage sur génome de référence avec BWA, le tout déployé sur un ordinateur classique, sur un cluster de calcul (PlaFRIM) et sur l’infrastructure GRISBI (900 processeurs et 26To de mémoire).
Globalement les résultats sont meilleurs que sur un ordinateur local mais moins bons que sur un (gros) cluster de calcul (local également) mais l’utilisation de la grille présente deux avantages :
– On peut déployer simultanément une quantité de processus en faisant varier un paramètre d’assemblage (du fait de la quantité de ressources disponibles et de la parallélisation massive des traitements)
– On peut accéder à cette architecture sans aucune contrainte matérielle au niveau local, ce qui est particulièrement le cas dans un laboratoire de biologie moléculaire.
Le travail présenté ici amène plusieurs perspectives, notamment la nécessité de rendre l’utilisation transparente à l’utilisateur final, biologiste, en utilisant,par exemple, un workflow (dans ce travail c’est Ergatis mais nous en reparlerons plus tard).
Au delà des résultats relativement préliminaires, on nous confirme que la mutualisation de ressources en général est une réponse efficace à ce type de problématique.

Une revue intéressante et qui se veut exhaustive sur les conséquences de la généralisation des technologies de séquençage et les solutions/adaptations possibles, on y retrouve pèle-mêle :
– Un listing à jour (2011) des différentes plateformes dédiées à la génération de données de séquençage (Illumina, Roche, Life Technologie pour ne citer qu’eux…) et leurs spécificités;
– La description de quelques stratégies de NGS : identification de variants, séquençage d’éxome, séquençage sur des régions précises…
– Les problématiques en bioinformatiques : stockage et analyse de données, développement de solutions logicielles adaptées…
– Les différentes analyses ainsi que des listes de logiciels pour répondre aux besoins: assemblage denovo et sur génome de référence, annotation et prédiction fonctionnelle, autant open-source que sous licence payante.
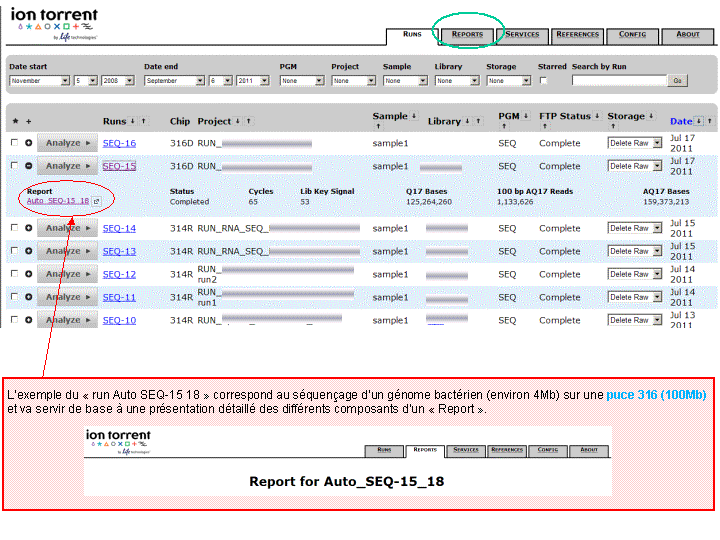
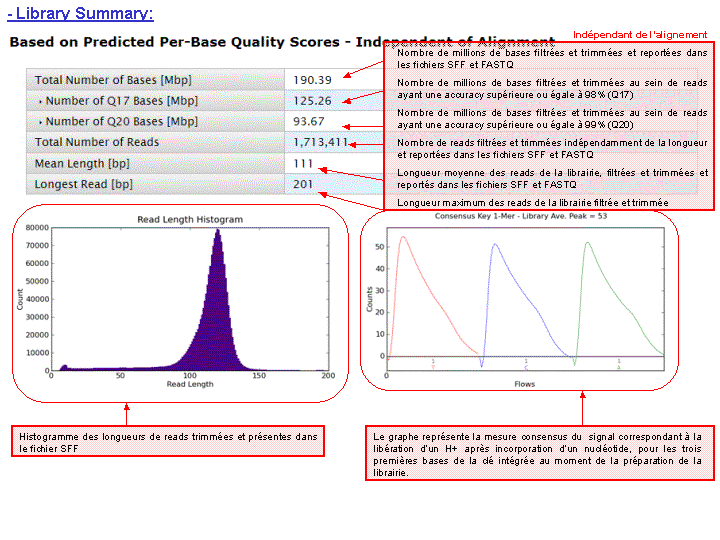
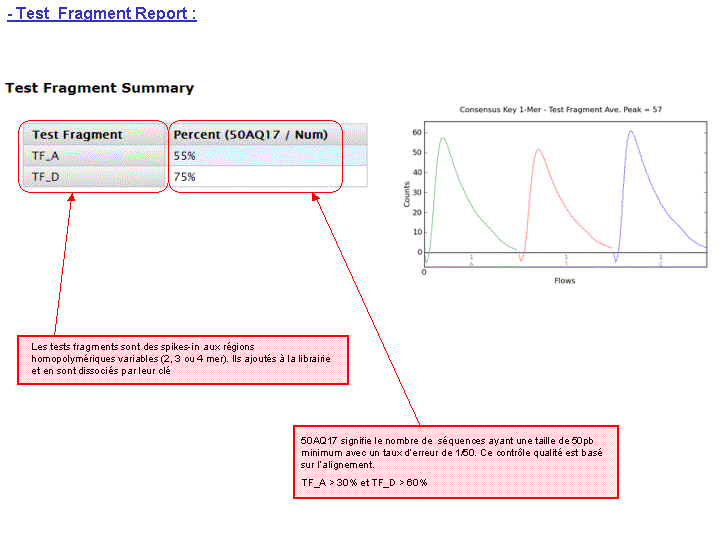
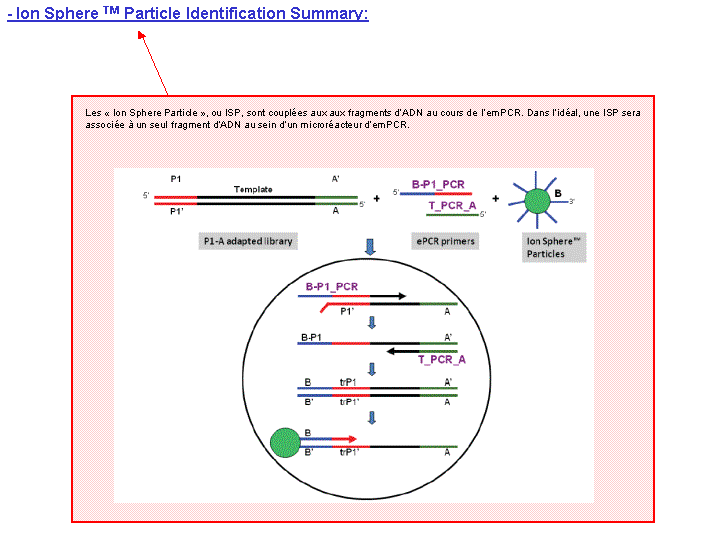
A l’issue d’un « run » de séquençage Ion Torrent (PGM), l’ensemble du signal brut (ionogramme) est converti en séquences et stocké au niveau du serveur. Pour chaque « run de re-séquençage», un alignement préliminaire est réalisé sur la base du génome de référence mentionné lors de l’initialisation du PGM. Cette information est reprise au travers d’un rapport qui comporte également un ensemble de paramètres, que l’on se propose de détailler ci-dessous :
(Cliquer pour agrandir) 
Le rapport se divise en 5 sections:

")

")
")





L’effervescence médiatique liée au séquençage de la bactérie Escherichia coli responsable de l’intoxication d’une dizaine de personnes dans le Nord de la France (juin 2011), a engendré de multiples reportages et communiqués. Parmi eux, celui de l’AFP, et ce paragraphe comme point de départ à cet article :
« L’Institut est équipé depuis un mois d’un séquenceur « à haut débit » qui permet de déchiffrer un génome en un temps record à partir d’une « librairie » d’ADN stockée sur une sorte de puce électronique. Cette technologie est notamment utilisée dans la recherche sur le cancer et pourrait remplacer dans un futur proche l’amniosynthèse (sic). »
En France, deux méthodes « invasives » permettent de réaliser un diagnostic prénatal : L’amniocentèse ou prélèvement du liquide amniotique, et une biopsie de trophoblastes ou prélèvement des cellules du futur placenta.
Toutefois, cet acte cause chaque année environ 1% de fausses couches. Rapporté aux 90000 amniocentèses par an, on déplore jusqu’à 900 pertes de fœtus, qui paradoxalement sont sain dans la majorité des cas.
Ces conséquences iatrogènes sont terribles sur le plan humain et il apparait évident de mettre au point une méthode beaucoup moins dangereuse pour la mère et surtout pour le fœtus.
Depuis plusieurs années, il a été démontré la présence de cellules d’origine fœtales dans le sang maternel dès la huitième semaine de grossesse. Cette particularité a dernièrement fait quelques émules et ont décidé de l’exploiter pour la mise au point d’un diagnostic prénatal « non invasif ». Le point critique repose sur la très faible proportion de ces cellules provenant du fœtus ; de l’ordre d’une pour dix millions de globules blancs et cinq milliards de glo¬bules rouges par millilitre de sang maternelle.
L’une des méthodes les plus encourageantes, réalisée par l’équipe de Patrizia Paterlini-Bréchot, Unité Inserm U807, repose sur une méthode d’enrichissement par la taille des cellules trophoblastique dans le sang, dite « ISET » ( Isolation by Size of Epithelial Tumor / Trophoblastic cells ), leur caractérisation par la technique des empreintes génétiques, puis séquençage de l’ADN extrait. La société « Rarecells » a par ailleurs était créée sur cette base.
Cette méthode sensible et spécifique à 100%, a déjà été validée techniquement et cliniquement pour le diagnostic prénatal non invasif de l’amyotrophie spinale (Beroud C., Karliova M., Bonnefont J.P., et al. Prenatal diagnosis of spinal muscular atrophy by genetic analysis of circulating fetal cells Lancet 2003 ; 361 (9362) : 1013-1014) et de la mucoviscidose ( « Recherche de la mutation du gène CFTR » Saker A., Benachi A., Bonnefont J.P., et al. Genetic characterisation of circulating fetal cells allows non-invasive prenatal diagnosis of cystic fibrosis Prenat Diagn 2006 ; 26 (10) : 906-916) , et techniquement seulement pour la trisomie 21.
Une seconde approche, réalisée par des chercheurs de l’Université de Stanford, est basée sur l’étude de l’ADN fœtal libre extrait du sang maternel puis analysé grâce aux séquenceurs à haut débit.
Dans le cas de la trisomie 21 par exemple, l’assignement de plusieurs dizaines de milliers de séquences présenterait un déséquilibre statistique même si la proportion d’ADN fœtal par rapport à l’ADN maternel est extrêmement faible (Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proceedings of the National Science Academy of the USA, 2008, 105, 16266–71). Si cette approche semble très attractive, plusieurs interrogations se posent suite à la lecture du travail scientifique, à la fois sur la méthodologie, l’analyse statistique et l’évaluation des résultats. (Commentaires sur les études de Fan et al.)
Dans l’éventualité d’un diagnostic basé sur le séquençage complet d’un génome, ou davantage ciblé tel l’ « amplicon sequencing » des gènes d’intérêts, la méthode non invasive s’appuyant sur une analyse globale du sang maternel nécessiterait une profondeur de séquençage conséquente. Ceci aurait pour incidence une orientation technique tournée vers des appareils de type « Solid 5500 » ou encore « Illumina HiSeq 2000 », cracheurs de « reads ».
Dans le cadre d’une application diagnostique, l’investissement financier est l’un des aspects considéré et en ce sens la méthode « ISET », plus contraignante dans la préparation du matériel biologique, permets néanmoins d’accéder, dans la perspective d’un séquençage ciblé, aux séquenceurs de paillasse, cinq à dix fois moins cher à l’achat et à des coûts de « runs » inégalés à ce jour. Record actuellement détenu par le PGM, Ion torrent dont la capacité des puces actuelles de 100Mb et d’1Gb à venir d’ici la fin d’année 2011, permettrait un multiplexage soit d’individus pour une mutation donnée, ou encore, imaginer une liste de séquences d’intérêts à diagnostiquer par individu.
Il y a fort à parier que le séquençage à haut débit finisse par devenir un outil incontournable dans le monde du diagnostic prénatal d’autant plus que les coûts d’équipement et de fonctionnement ne cessent de diminuer. Dans cette perspective, l’évolution vers les séquenceurs haut-débit de 3ème génération (à partir de cellule unique) pourrait faire la part belle à l’utilisation des cellules trophoblastiques.
Au travers de ces avancées et de l’ère du séquençage à haut débit, les perspectives d’un diagnostic prénatal non invasif semblent se dessiner et laisse entrevoir un énorme soulagement. Aussi, la possibilité de mettre en place des plates-formes permettant de cribler l’essentiel des pathologies génétiques est envisagée.
Aussitôt, les détracteurs soulèvent la notion d’eugénisme, dénonçant un diagnostic prénatal intégral en vue d’une « traque » des individus « non conformes ».
Il apparait évident que ces avancées technologiques risquent très prochainement de soulever de réels débats de société…
Depuis 2005, le séquençage haut débit, comme son nom l’indique, a permis d’accroître la quantité de séquences produites par unité de temps, d’individu et de machine. Si intrinsèquement le terme de révolution est associé à ce type de technologies, il semble qu’il serait plus indiqué de l’associer aux nouvelles approches qui en découlent.
En effet, le séquençage nouvelle génération permet d’aborder des études sous de nouveaux angles d’approches. Bien souvent ces approches existaient pour la plupart avant l’avènement de ces nouvelles machines mais leur mise en œuvre étaient bien souvent laborieuses, coûteuses. Beaucoup de techniques nécessitaient des a priori techniques ou scientifiques (des a priori dus à la sélection et aux designs de sondes déposées ou synthétisées sur un support solide dans le cas des puces à ADN permettant les études transcriptomiques). Les nouvelles méthodes de séquençage, quant à elles, permettent de lever certaines anticipations expérimentales. Ainsi une étude du niveau de modulation des transcrits peut grâce à l’emploi de ces technologies en découvrir de nouveaux, ce que ne permet pas ce même type d’études sur puces à ADN. En outre tout a priori constitue un biais expérimental potentiel.
Pour résumer, un peu simplement, le séquençage haut débit dépasse l’outil analytique pour devenir une méthode exploratoire à part entière.
L’objectif de cet article est de proposer un bref aperçu du spectre d’applications et des champs d’expertises que ces nouvelles approches révolutionnent (nous reviendrons plus tard plus en détail sur certaines).
Concernant les applications ayant pour finalité les études génomiques, sont à distinguer :
– Séquençage de novo
Cette application découle de la quantité même de séquences que ces nouvelles générations de machines sont capables de générer. Aujourd’hui il est admis qu’un séquençage de novo nécessite une profondeur de 25 X, c’est-à-dire qu’il est possible de séquencer l’ADN d’un organisme procaryote en un run de séquençage sur la plupart des configurations de séquenceurs. Ce type d’applications a pleinement bénéficié des outils bio-informatiques au niveau des logiciels, machines et compétences humaines de plus en plus disponibles pour tenter de banaliser cette application. Ainsi, le centre de Shenzen avec le BGI (Beijing Genomics Institute, Chine) propose deux projets (pompeusement intitulés library of digital life) le premier consiste au séquençage (et reséquençage) de 1000 génomes de plantes et d’animaux et de 10 000 génomes microbiens.
– Découvertes de SNPs (Single Nucleotide Polymorphisms)
Cette application a très vite trouvé une application directe, elle a contribué au développement de puces à ADN de génotypage haut débit. Ainsi Illumina a pu produire en quelques mois des puces à ADN permettant le génotypage en parallèle d’environ 2,5 millions de SNPs par échantillon (un format 5 millions de SNPs est en préparation) en s’appuyant sur les résultats du consortium 1000 génomes qui a enrichi les bases de données en variations génétiques de la séquence consensus humaine. Ces outils permettant de réaliser des études d’association en réalisant des profils génétiques de plus en plus résolutifs.
Pour ce type d’application, deux modalités sont à distinguer :
– le reséquençage ciblé de zones d’intérêt, étape faisant suite, par exemple, à une étude d’association et permettant après reséquençage d’un locus génétique associé à un caractère particulier, de déterminer la causalité du phénotype différentiellement observé en terme de séquence.
– le réséquençage exhaustif d’ADN génomique. Cette modalité quant à elle, permet la mise en œuvre d’études de comparaisons génétiques de souches (telle que le permet la CGH (Comparative Genomic Hybridization) en s’affranchissant de toute hybridation grâce au séquençage direct)
Sous ce champ expérimental des études génomiques, peuvent être classées toutes les études de métagénomiques où un milieu cherche à être caractérisé le plus exhaustivement et finement possible par la diversité et le degré de contribution de chaque micro-organisme vivant (ou mort…) qui le compose. Le séquençage haut débit permet de rendre accessibles ce type d’approches.
Etudes transcriptomiques
Des méthodologies employées sur puces à ADN telles que les études transcriptomiques ont évolué et été adaptées sur les plateformes de séquençage haut-débit. En outre, ces nouvelles approches permettent de mesurer plus finement des niveaux de modulation tout en tenant compte des isoformes des transcrits. Encore une fois, le fait de séquencer permet de limiter les biais en comparaison de l’emploi de puce à ADN. La lecture plus directe s’affranchit des éléments de design de sondes, des phénomènes d’hybridation etc. A terme, les séquenceurs haut débit supplanteraient les puces à ADN pour ce qui concerne les applications d’études transcriptomiques.
– Réalisation de profils d’expression globale où l’intégralité du transcriptome cherche à être finement caractérisé pour une condition donnée.
– Caractérisation d’ARN non codant
Lors d’études de profils d’expression, les méthodes de séquençage haut débit permettent d’envisager la détermination et caractérisation des ARNs non codants (ici sont particulièrement visés les miRNA et smallRNA).
Etudes épigénétiques
– Etudes de la méthylation de l’ADN (méthyl-seq)
Les études de la méthylation de l’ADN génomique cherche à cartographier les loci fortement métyhylés dans une circonstance donnée. Pour rappel, une faible méthylation favorise la transcription mais une forte méthylation, au contraire, l’inhibe. Lorsque le promoteur d’un gène est méthylé, le gène en aval est réprimé et est donc plus difficilement ou pas du tout transcrit en ARNm.
– Etudes d’association protéines-ADN
Le ChIP-séquençage, également connu sous l’appellation de ChIP-Seq, est utilisé pour analyser des interactions protéines/ADN. Le ChIP-Seq combine immunoprécipitation des zones génétiques sur lesquelles se trouvent fixées des protéines (ChIP) avec le séquençage haut débit de l’ADN afin d’identifier des motifs consensus. Il peut être utilisé pour une cartographie précise de sites de liaison pour une protéine d’intérêt.
Ces deux dernières applications ont dans un premier temps été développées sur la base des tiling-arrays. Le séquençage haut débit permet de diminuer les coûts d’investigation tout en gagnant en sensibilité.
Des technologies émergentes permettent souvent d’envisager de nouvelles applications diagnostiques. Ainsi quelques études depuis 2008, (Fan et al., Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proceedings of the National Science Academy of the USA, 2008, 105, 16266–71), semblent ouvrir la porte à un diagnostic prénatal non invasif.
La recherche médicale avec son débouché clinique de la médecine personnalisée entrevoit des applications au séquençage haut débit. Ainsi, une équipe, montre dans des résultats publiés dans Nature Genetics, pour la troisième fois au monde, la faisabilité d’une étude menée sur la base du séquençage de l’exome, aboutissant à la découverte d’une causalité génétique (simple, puisque monogénique). Cette mutation du gène NOTCH2 causalité d’une maladie rare, le syndrome de Hadju Cheney, une ostéoporose sévère, a été identifiée et caractérisée efficacement par l’application des techniques de séquençage haut débit. Il y a peu ce type d’identifications n’auraient pas été financées puisque trop longue à mener, trop coûteuses pour des retombées certainement perçues comme limitées. Pour beaucoup, ces études menées à grandes vitesses trouveront des applications concrètes dans le champ de la médecine personnalisée… mais de cela nous reparlerons.

Le séquenceur de paillasse PGM IonTorrent
Notre laboratoire possède un PGM IonTorrent de Life Technologies. Cet outil haut-débit est un séquenceur de paillasse permettant de réaliser des runs à prix relativement accessible, inférieur à 800€/run (cf. notre historique du séquençage proposé dans un post précédent).
Nous avons pu réaliser, en avant-première, un séquençage complet d’une souche d’Escherichia coli sur une puce 316 ,qui permet d’obtenir 100Mb / run, soit l’équivalent d’un peu plus d’une couverture de 20X d’une Escherichia coli.
Pour plus d’informations sur ce séquençage, vous pouvez lire:
– le communiqué de presse associé : CP_Escherichia_coli
-L’article de Futura Sciences sur le sujet : http://www.futura-sciences.com/fr/news/t/genetique-1/d/sequencage-haut-debit-de-coli-vers-une-medecine-personnalisee_31316/
– Des articles plus généralistes sortis dans différents journaux :
Ou regarder ces reportages (datés du 06/07/2011) :
Reportage-LCI-séquençage-coli-06072011
Reportage-BFMTV-séquençage-coli-06072011

10 chips 314 = 1 chip 316

étape de préparation de la matrice enrichie
Le procédé de séquençage se déroule sur 3 jours :
– préparation de la librairie : 1 jour (fragmentation de l’ADN génomique…)
– préparation de la matrice : 1 jour (PCR en émulsion + enrichissement)
-séquençage de la matrice enrichie : 1/2 jour

c'est parti !
Et on obtient le report suivant : Report Run IonTorrent
(*) Sequencing data were generated using system software and protocols bothnon released and non supported by Ion TorrentTM (part of Life TechnologiesTM) and may not reflect actual Ion Torrent PGMTM performance in term of throughput and/or accuracy.
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.


{kind=link}
{kind=link}
{kind=link}
{kind=link}