
Currently viewing the category:
"Comparatif"
Le choix d’une méthode analytique confortable, fiable, adaptée au profil de données (avec un peu ou beaucoup d’erreurs liées à la technologie de séquençage utilisée), à votre degré d’expertise, est souvent un casse-tête et un dédale qui occupent certains partenaires d’un projet nécessitant le recours à une analyse métagénomique ciblée. D’autres encore décident de ne pas décider, de ne pas se lancer dans le labyrinthe et votent ainsi pour une solution tout-terrain, sorte de martingale génomique, proposée par des prestataires de services.
Cet article de Plos One : Assessment of Common and Emerging Bioinformatics Pipelines for Targeted Metagenomics tente – ayant un conflit d’intérêt majeur, je n’en ferai pas la critique – d’identifier les grandes approches algorithmiques disponibles. Quels sont les paramètres influençant la qualité des résultats ?
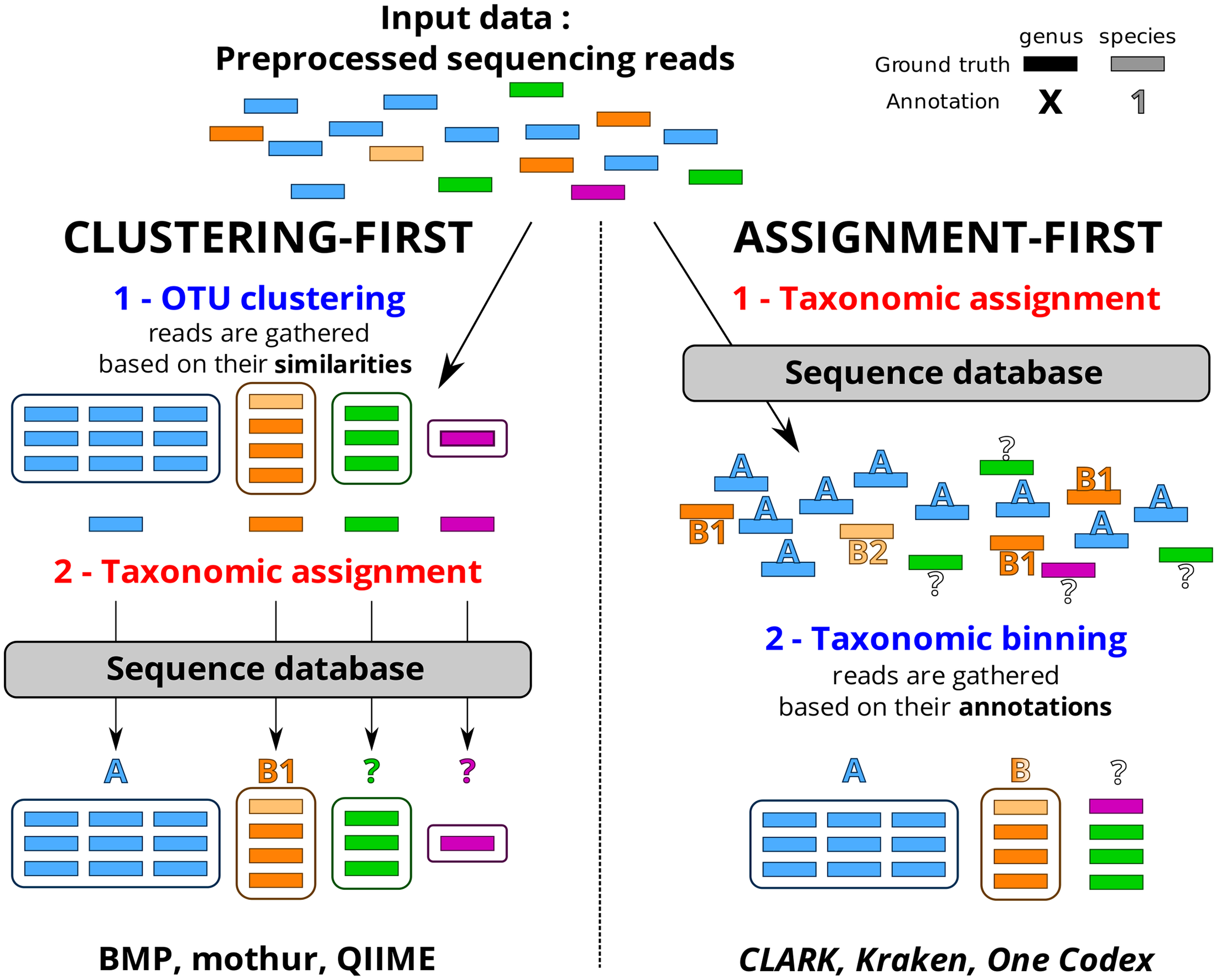
Ce travail a permis de fournir un canevas généralisable permettant d’évaluer un pipeline analytique et d’observer dans quelle mesure ce dernier influe sur la qualité des résultats. Concernant le protocole mis en place ainsi que d’autres ressources nécessaires (jeu de données simulées, réelles etc.) vous pourrez consulter la page dédiée à ce travail sur le site de PEGASE-biosciences.
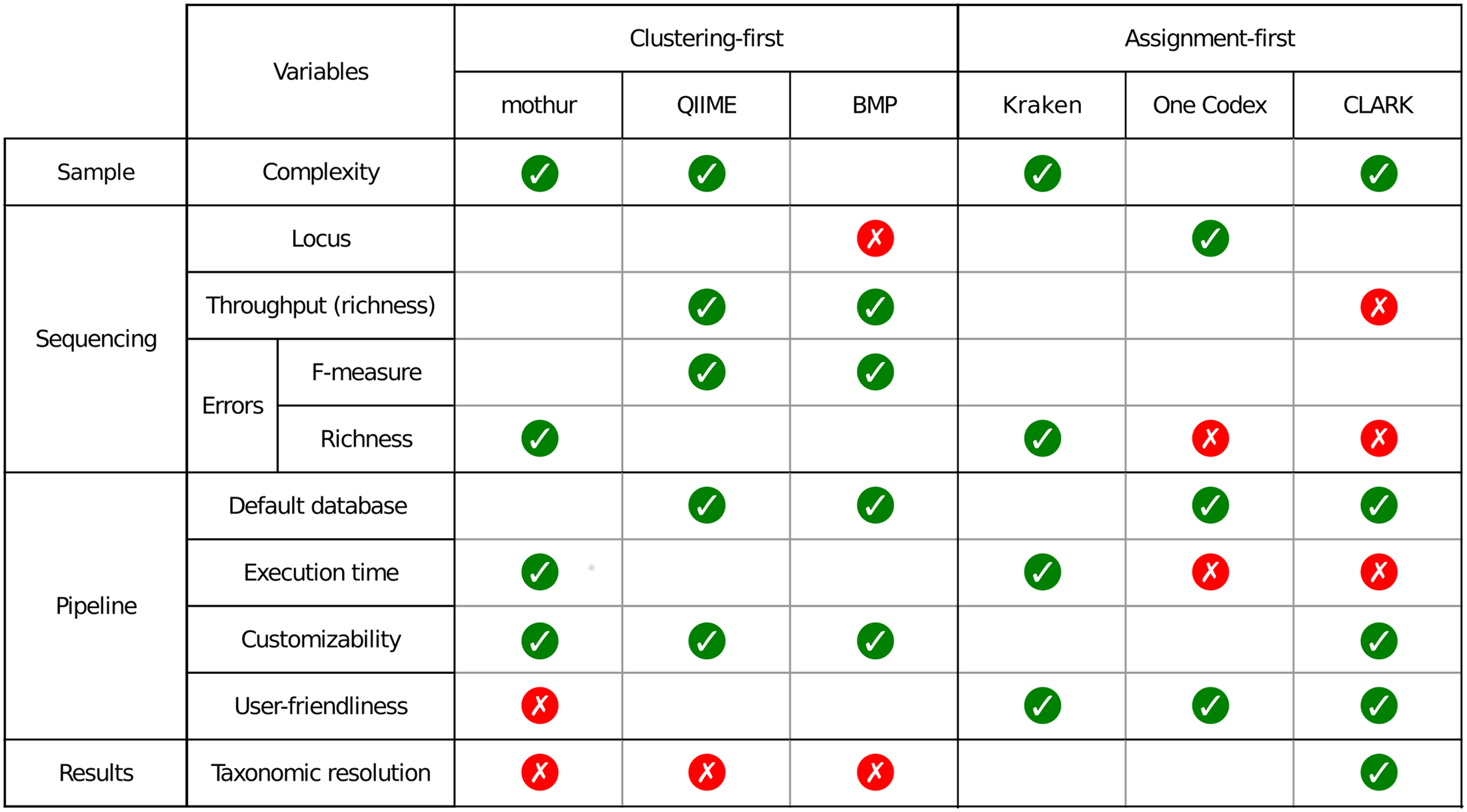
Un petit tableau synoptique permet de synthétiser les caractéristiques principales des pipelines évalués dans l’article.
 Le séquençage haut-débit utilisant des analogues de nucléotides « portant des groupes bloquants en 3′ qui sont utilisés pour produire une terminaison de chaîne réversible appliqués pour le séquençage par synthèse nucléotidique » (je sais cette phrase est « un peu » lourde…) est un brevet de Caliper Technologies -autrement dit de Perkin Elmer : la firme qui n’a pas su saisir sa chance lors de la révolution du séquençage haut-débit. Le séquençage par synthèse (avec réversion du blocage des nucléotides fluorescents polymérisés -un peu moins lourde cette phrase là…) est le cœur de la technologie faisant une partie du chiffre d’affaire d’Illumina (l’intégralité de sa gamme de séquenceurs fonctionne sur ce principe).
Le séquençage haut-débit utilisant des analogues de nucléotides « portant des groupes bloquants en 3′ qui sont utilisés pour produire une terminaison de chaîne réversible appliqués pour le séquençage par synthèse nucléotidique » (je sais cette phrase est « un peu » lourde…) est un brevet de Caliper Technologies -autrement dit de Perkin Elmer : la firme qui n’a pas su saisir sa chance lors de la révolution du séquençage haut-débit. Le séquençage par synthèse (avec réversion du blocage des nucléotides fluorescents polymérisés -un peu moins lourde cette phrase là…) est le cœur de la technologie faisant une partie du chiffre d’affaire d’Illumina (l’intégralité de sa gamme de séquenceurs fonctionne sur ce principe).
Le principe assez simple du séquençage SBS (Sequencing By Synthesis) est en train de se décliner au sein de machines haut-débit plus accessibles en terme de coût… il s’agit là de l’arrivée sur le marché d’une nouvelle concurrence : des séquenceurs génériques tels que ceux que proposent le distributeur Azco Biotec, Inc. Ces séquenceurs viennent directement concurrencer ceux d’Illumina accompagnés de leurs réactifs « génériques » tels que ceux distribués par la société Proteigene…
Ces séquenceurs « génériques » ne révolutionnent rien, leur part de marché est actuellement marginale… les laboratoires académiques préfèrent souvent l’original à la copie, malgré tout la concrétisation de la « promesse » –qui l’a formulée au juste ?– d’un génome humain pour 100 $ d’ici 2015 passe par la large diffusion de machines dont la technologie a fait ses preuves… Le retard pris par les séquenceurs de 3ème génération laisse le champ libre à la 2ème. Le marché risque de se saturer en séquenceurs de paillasse (séquenceur de 2ème génération tels que le MiSeq d’Illumina et les PGM et Ion Proton de Life Technologies).

Face aux arguments commerciaux développés en faveur de tels ou tels séquenceurs de paillasse à haut débit, l’étude menée par Loman et al., Nature Biotechnology propose une comparaison des performances du 454 GS Junior (Roche), du MiSeq (Illumina) et du PGM Ion torrent (Life Technologies), à partir du séquençage de novo d’un isolat d’E.coli O104 :H4 responsable de l’intoxication alimentaire en Allemagne (Mai 2011). Pour cela, le génome de référence a été généré à l’aide du GS FLX (Roche) et permettra d’évaluer l’efficacité des séquenceurs et algorithmes d’assemblages associés.
Sur la base des caractéristiques techniques établies en 2011, les trois plateformes ont permis de générer un draft exploitable du génome bactérien sans pour autant parvenir à le reconstituer à 100%. Par conséquent, il est impossible de désigner un séquenceur comme étant celui de choix, chaque plateforme étant un compromis entre avantages et inconvénients.
Deux « runs » de 454 GS Junior (associés au bénéfice de reads de 600 bases) ont permis d’atteindre 96,28% de couverture par rapport au génome de référence (Contre 96,05% pour le MiSeq et 95,4% pour le PGM Ion torrent).
Malgré une capacité de séquençage du PGM Ion torrent supérieure à celle du 454 GS Junior (100Mb contre 70Mb) et une précision de séquençage voisine (Q20, soit 1 erreur toutes les 100 bases), la technologie reposant sur les semiconducteurs fait face à ses limites au niveau de la gestion des homopolymères et accessoirement d’une longueur de reads de 100 bases.
Vingt fois supérieur au 454 GS Junior en terme de quantité de bases décodées, un « run » de MiSeq ne parvient qu’à talonner les performances d’un double « run » de la technologie Roche. Bien qu’étant la seule à gérer les homopolymères et la seule doter d’une précision à Q30 (soit 1 erreur toutes les 1000 bases), la technologie Illumina faillit sur ses tailles de « reads » qui ne permettent pas une gestion optimum des séquences répétées lors de l’assemblage.
L’approche analytique est également évoquée et selon le système d’assemblage utilisé (Velvet, CLC, MIRA), de nombreuses différences sont observées.
Enfin, ce document est l’occasion de rassembler des notions assez pragmatiques comme le tarif des plateformes (et annexes associées), la durée d’un « run » de séquençage ou encore la quantité de données générées.
Ceci étant, on est en droit d’émettre quelques réticences quant à la pertinence de cette étude qui base son comparatif sur une application (séquençage de novo) qui n’est pas la vocation première de cette catégorie de séquenceurs. Par ailleurs, dans le laps de temps où cette étude a été réalisée et la parution des résultats, la technologique du PGM Ion torrent a évolué. Testé sur les bases d’une puce 316 (100Mb) et d’une technologie de 100 bases en longueur de « reads », le PGM bénéficie désormais de la puce 318 (1 Gb) associée à une technologie de reads de 200 bases. Sans tarder, Life technologies réagit (“Loman et al reflects the past, not the present”) et propose une mise à jour au travers de ce document.

La confusion entre mate-pair et paired-end, tant au niveau technologique (selon qu’on lise les notes techniques d’Illumina, de Roche ou de Life) que logiciel nous a mené à rédiger, en collaboration avec Ségolène Caboche, Bioinformaticienne à l’université de Lille2, une note technique dont le contenu est résumé ci-dessous :
– Genèse de la confusion entre mate-pair et paired-end
– Descriptions les deux approches, avec un focus sur les principales technologies de seconde génération de séquenceurs
– Traitement au niveau logiciel et conseils généralistes pour l’utilisation
Le document est consultable dans son intégralité sur notre blog :
Télécharger Paired-end versus mate-pair
Bonne lecture!
Ce post fait naturellement suite à celui dédié à la seconde génération de séquenceurs multi-parallélisés, et conserve la même approche, à savoir un tour d’horizon des technologies et une évocations des informations générales sur le sujet.
A l’instar du PGM de Ion torrent mis sur le marché depuis un an (10Mb – reads 100b – 06.2011 / 100Mb – reads 200b -11.2011 / 1Gb – reads 400b – prévu début 2012), la seconde génération de séquenceurs haut débit tend vers une production de reads de plus en plus longs et de moins en moins chère. Toutefois, on est en droit de se demander quelle sera leur pérennité face à la 3éme génération répondant à un cahier des charges assez similaire et la possibilité de bénéficier de nouvelles applications.

Le principe de la 3ème génération peut être symbolisé par le séquençage d’une molécule d’ADN sans étape de pré-amplification (contrairement à la génération actuelle type 454 Roche, SOLiD Life technologie, Ion Proton, PGM Ion torrent, HiSeq Illumina, …) en conservant l’incorporation de nucléotides, par cycles ou non ( dans ce dernier cas, le terme de « Séquençage d’ADN simple molécule en temps réel » est approprié).
Les technologies « SMS » pour « Single Molecule Sequencing » peuvent être regroupées selon trois catégories:
– Technologies de séquençage en temps réel impliquant la synthèse du brin d’ADN complémentaire via une ADN polymérase.
– Technologies de séquençage par détection des bases successives d’une molécule d’ADN au travers de nanopores.
– Technologies de séquençage basées sur des techniques de microscopie.
En combinant les dernières avancées dans la nanofabrication, la chimie de surface et l’optique, Pacific Biosciences (Pacbio RS) a lancé une plateforme technologique puissante appelée technologie de molécule unique en temps réel, ou « SMRT » pour « Single Molecule Real-time sequencing ». Parmi ses concurrents directs, Helicos Biosciences (Helicos) qualifié « tSMS » pour « True Single Molecule Sequencing ». Malgré le recours à une technologie analogue, la mention « Temps réel » auquel il échappe est simplement liée à une incorporation cyclique des nucléotides fluorescents.
D’autres technologies, à des degrés de développement plus ou moins avancé, sont dans les tuyaux et qui sait de Noblegen, Starlight, Cracker Bio, NABSys, Halcyon, ou autres… révolutionnera encore un peu plus cet univers du haut débit et suivra le chemin emprunté dernièrement par Oxford Nanopore …

Le terme « Nouvelle génération de séquençage à haut-débit » ( ou « Next generation sequencing » ) regroupe l’ensemble des technologies ou plateformes de séquençage développées depuis 2005 par quelques sociétés de biotechnologies.
L’objectif de cet article est de proposer de manière synthétique, un tour d’horizon des différents principes et caractéristiques de ces nouveaux outils et ainsi fournir quelques orientations et solutions techniques en réponses à des questions biologiques.
La position actuelle dans laquelle nous nous trouvons, entre la commercialisation de certains séquenceurs et ceux en cours de développement, est caractéristique d’une période charnière dissociant les technologies à haut débit dites de 2ème génération qui requièrent une étape d’amplification des molécules d’ADN en amont du décodage, de celles dites de 3ème génération permettant le décryptage direct d’une seule molécule d’ADN. Cette dernière catégorie fera à elle seule l’objet d’un prochain article.
Le marché des séquenceurs de 2éme génération est couvert par 3 grand groupes que sont Roche, Illumina et Life Technologies ayant respectivement proposés de manière successive, leur première plateforme à savoir le 454, le Genome Analyser et enfin le SOLiD. Depuis, le marché s’est étoffé proposant un panel de technologies au principe et caractéristiques propres telles qu’elles sont mentionnées ci dessous. A noter que parmi ce panel, le PGM, Ion torrent est le seul a connaitre une évolution constante en terme de capacités de séquençage (10Mb – reads de 100b – Juin 2011 / 100Mb – reads de 100b – Sept 2011 / 100Mb – reads 200b – Nov 2011 / 1Gb – Jan 2012 )

Chaque plateforme possède ses avantages et inconvénients et nombreuses sont celles configurées pour répondre à de nombreuses approches « omics », dans certaines limites. Il s’agira de faire un choix technologique selon les champs d’applications souhaités.
De manière générale, le type d’organisme étudié prédéterminera la technologie à employer. La notion de profondeur est récurrente à chaque application et dans l’objectif d’un reséquençage, le choix de la plateforme peut être identifié, de manière simplifiée, sur la base d’un calcul rapide ( P = N / L où P: Profondeur, N: Nombre des nucléotides totaux des reads, L: Taille du génome étudié).
Concernant les séquenceurs de 2ème génération, le séquençage de novo est une application mentionnée chez de nombreux fournisseurs (cf le tableau ci-dessous). Toutefois, l’association de deux technologies générant à la fois des reads longs (type 454, Roche) et une profondeur conséquente (type GAIIx, Illumina) palliant aux problèmes liés aux homopolymères et erreurs de séquençage, est préconisée (Au cours de l’article à venir sur les séquenceurs de 3ème génération, nous aborderons les plateformes davantage configurées pour cette application).
Ce paramètre de profondeur sera également à prendre en considération pour les champs d’applications incluant la notion d’analyse quantitative (RNAseq, ChIPseq, …). Si la profondeur permet d’atténuer les erreurs de séquençage, il reste néanmoins préférable de s’orienter vers des technologies à Q30 minimum (1 erreur sur 1000) pour la détection de SNPs.

Selon les technologies évoquées ci-dessus, les caractéristiques et champs d’applications ont évolués. Aussi, je vous propose de retrouver l’ensemble de ces informations actualisées en cliquant sur ce lien.
L’ensemble des informations sont détaillées dans l’article mentionné ci-dessous:

Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

