
Currently viewing the tag: "ngs"
Ce billet très court, pour faire l’annonce (un peu tardive) du début d’un MOOC. Ce dernier s’annonce excellent, traitant du séquençage de génomes complets bactériens.

Ce cours estival est proposé par l’excellente Université Technique du Danemark (DTU)
En voici le plan :
WEEK 1
Module 1
Welcome and introduction to typing of bacteria and use of Whole genome sequencing applied to surveillance of bacterial pathogens and antimicrobial resistance
3 vidéos
WEEK 2
Module 2
Introduction to Next Generation sequencing
3 vidéos
WEEK 3
Module 3
Whole genome sequencing tools- demonstration of analysis tools for species identification, MLST typing and finding resistance genes
3 vidéos
WEEK 4
Module 4
Whole genome sequencing tools- demonstration of analysis tools for Serotyping of Salmonella and Escherichia coli strains , and finding plasmid replicons
3 vidéos
WEEK 5
Module 5
Whole genome sequencing tools- demonstration of analysis tools for multiple analyzes, phylogenetic tree building and finding genetic markers from self-made databases and Summative Tutorial exercise
5 vidéos, 1 lecture
Noté: Tutorial final Quiz
 L’accélération du débit bibliographique faisant référence aux « miRNA » atteste aisément de leur caractérisation récente (Lee RC et al., Cell (1993)) et de l’intérêt lié à leurs potentielles fonctions .
L’accélération du débit bibliographique faisant référence aux « miRNA » atteste aisément de leur caractérisation récente (Lee RC et al., Cell (1993)) et de l’intérêt lié à leurs potentielles fonctions .
Il aura fallu près de dix années supplémentaires pour mettre en évidence leur implication en tant que régulateurs biologiques (notamment au niveau de la régulation de l’expression des gènes) et leurs impacts dans de certains cancers… Aussi, le développement des nouvelles technologies de séquençage à haut débit contribue forcément à cette émergence.
Ce poste est l’occasion de présenter « miRNAtools » qui comme son nom l’indique, regroupe un grand nombre de liens renvoyant vers différents outils dédiés aux miRNA.

– Analyse de données NGS appliquées aux miRNAs (étude des profils d’expression). La liste des 7 softwares présentés n’est pas exhaustive et en voici quelques uns supplémentaires à tester: « mireap », « miRTRAP », « DSAP », « mirena », « miRNAkey », « SeqBuster », « E-mir », … . Une comparaison de l’efficacité de certains de ces outils fera l’objet d’un prochain poste.
– Prédiction de cibles selon les miRNA étudiés.
– Analyse de pathways impliquant les miR d’intérêt. Pour cette dernière application, le soft DIANA LAB – Mirpath proposé, bien que facile d’utilisation et gratuit, a le défaut de ne s’appliquer qu’aux organismes « humain » et « souris ». Dans ce registre et moyennant quelques milliers d ‘euros, « Ingenuity Pathway Analysis » (« IPA ») reste de loin l’outil idéal. En effet, en plus d’identifier les voies métaboliques au sein desquels sont impliqués les miR modulés comme proposé par Mirpath, « IPA » permet également d’intégrer les résultats de modulation de miR et d’expression de gènes pour une même condition d’étude…
La confusion entre mate-pair et paired-end, tant au niveau technologique (selon qu’on lise les notes techniques d’Illumina, de Roche ou de Life) que logiciel nous a mené à rédiger, en collaboration avec Ségolène Caboche, Bioinformaticienne à l’université de Lille2, une note technique dont le contenu est résumé ci-dessous :
– Genèse de la confusion entre mate-pair et paired-end
– Descriptions les deux approches, avec un focus sur les principales technologies de seconde génération de séquenceurs
– Traitement au niveau logiciel et conseils généralistes pour l’utilisation
Le document est consultable dans son intégralité sur notre blog :
Télécharger Paired-end versus mate-pair
Bonne lecture!
Ce post fait naturellement suite à celui dédié à la seconde génération de séquenceurs multi-parallélisés, et conserve la même approche, à savoir un tour d’horizon des technologies et une évocations des informations générales sur le sujet.
A l’instar du PGM de Ion torrent mis sur le marché depuis un an (10Mb – reads 100b – 06.2011 / 100Mb – reads 200b -11.2011 / 1Gb – reads 400b – prévu début 2012), la seconde génération de séquenceurs haut débit tend vers une production de reads de plus en plus longs et de moins en moins chère. Toutefois, on est en droit de se demander quelle sera leur pérennité face à la 3éme génération répondant à un cahier des charges assez similaire et la possibilité de bénéficier de nouvelles applications.

Le principe de la 3ème génération peut être symbolisé par le séquençage d’une molécule d’ADN sans étape de pré-amplification (contrairement à la génération actuelle type 454 Roche, SOLiD Life technologie, Ion Proton, PGM Ion torrent, HiSeq Illumina, …) en conservant l’incorporation de nucléotides, par cycles ou non ( dans ce dernier cas, le terme de « Séquençage d’ADN simple molécule en temps réel » est approprié).
Les technologies « SMS » pour « Single Molecule Sequencing » peuvent être regroupées selon trois catégories:
– Technologies de séquençage en temps réel impliquant la synthèse du brin d’ADN complémentaire via une ADN polymérase.
– Technologies de séquençage par détection des bases successives d’une molécule d’ADN au travers de nanopores.
– Technologies de séquençage basées sur des techniques de microscopie.
En combinant les dernières avancées dans la nanofabrication, la chimie de surface et l’optique, Pacific Biosciences (Pacbio RS) a lancé une plateforme technologique puissante appelée technologie de molécule unique en temps réel, ou « SMRT » pour « Single Molecule Real-time sequencing ». Parmi ses concurrents directs, Helicos Biosciences (Helicos) qualifié « tSMS » pour « True Single Molecule Sequencing ». Malgré le recours à une technologie analogue, la mention « Temps réel » auquel il échappe est simplement liée à une incorporation cyclique des nucléotides fluorescents.
D’autres technologies, à des degrés de développement plus ou moins avancé, sont dans les tuyaux et qui sait de Noblegen, Starlight, Cracker Bio, NABSys, Halcyon, ou autres… révolutionnera encore un peu plus cet univers du haut débit et suivra le chemin emprunté dernièrement par Oxford Nanopore …

Il y a quelques semaines, nous avions discuté de l’utilisation de logiciel de Workflow pour la bioinformatique. Il est temps de passer à la pratique en vous présentant un de ses dignes représentants : Galaxy.

Page d'accueil du site du workflow Galaxy
Le workflow Galaxy fournit un ensemble d’outils pour la manipulation et l’analyse de données génomiques. Il est très intuitif dans l’utilisation ce qui en fait une cible de choix pour le biologiste.
Il est possible d’utiliser Galaxy directement depuis le serveur. Avantage conséquent pour les bioinformaticiens il est possible d‘installer sa propre instance de serveur Galaxy, cette option fera l’objet d’un prochain post technique.
Du point de vue de l’interface graphique :

Interface principale - Workflow Galaxy
On peut également créer des workflows, les enregistrer dans un espace dédié, les partager, et les exécuter de façon automatique.
Pour exemple ce workflow de métagenomique publié gratuitement par un utilisateur de Galaxy (vous devez être connecté pour visualiser le workflow dans Galaxy)

Workflow analyse métagénomique - Galaxy
Les outils dédiés analyse de données NGS sont régulièrement mis à jour et nul doute que d’ici peu, certains seront dédiés IonTorrent.
Les tutoriels sont également très bien faits, on apprend très vite à maitriser l’environnement grâce à des dizaines de vidéos d’aides.
Galaxy offre donc la possibilité d’exécuter des analyses bioinformatiques sans effort de programmation. La version en ligne est intéressante car elle permet de se familiariser aux logiciels et d’exécuter l’analyse depuis un portable, mais la possibilité d’intégrer ces propres outils (nous y reviendrons) est indéniablement un gros avantage de la version locale.
Si nous devions citer un inconvénient, plutôt d’actualité : l’utilisateur est obligé de charger ses données en mémoire dans Galaxy, le temps de chargement peut être très long si l’on manipule des données issues d’expériences NGS. D’autres workflows tels que Ergatis, fonctionnent en local et permettent à l’utilisateur d’utiliser directement les données présentent sur l’ordinateur.
Pour en savoir plus :
La description complète du logiciel Galaxy en Français sur PLUME :
http://www.projet-plume.org/fiche/galaxy
Le Galaxy Wiki :
http://wiki.g2.bx.psu.edu/FrontPage
La publication associée :
![]()
Voici un excellent rapport (toujours d’actualité) réalisé par J.P. Morgan, une holding leader dans la banque d’investissement. Ce rapport traite de la part prise par les différentes applications NGS, il compare les différents séquenceurs haut-débit… cette enquête a été réalisée en interrogeant 30 laboratoires (dont 24 Américains), 50 % de ces laboratoires sont publics. ..

Ce rapport permet d’entrevoir ce que seront les prochaines demandes de financements des laboratoires d’une part, d’autre part, il permet d’entrevoir les possibilités offertes par ce type de technologies encore très consommatrices de fonds et de temps humain. Les technologies disponibles sont de plus en plus diverses… Pour beaucoup de laboratoires il s’agira d’effectuer le choix technologique le plus adapté à leur champ d’application. Avec un petit étonnement, on apprend que l’application la plus développée au sein des laboratoires, concerne l’expression de gènes (mRNA expression profiling)… le séquençage de novo n’arrivant qu’en 7ème place des applications les plus développées.
(le rapport est disponible en cliquant sur l’image ci-dessus)
Le PPF Bioinformatique de l’Université Lille 1, l’IFR 142 Médecine Cellulaire et Moléculaire de l’Institut Pasteur de Lille et l’IFR 114 Médecine Prédictive et Recherche Thérapeutique organisent une journée scientifique sur le thème de l’analyse bio-informatique des données produites par les technologies de séquençage à haut débit, dans la lignée de l’édition de 2009. Cette année, les sujets abordés couvriront plus particulièrement les données de type RNA-seq, ChIP-seq,…
Les exposés se tiendront dans l’amphithéatre Butiaux, sur le campus de l’Institut Pasteur de Lille (métro ligne 2, station Grand Palais).
Cette journée sera jumelée au workshop Algorithmique, combinatoire du texte et applications en bio-informatique, qui aura lieu à partir du lendemain au même endroit.

Une revue intéressante et qui se veut exhaustive sur les conséquences de la généralisation des technologies de séquençage et les solutions/adaptations possibles, on y retrouve pèle-mêle :
– Un listing à jour (2011) des différentes plateformes dédiées à la génération de données de séquençage (Illumina, Roche, Life Technologie pour ne citer qu’eux…) et leurs spécificités;
– La description de quelques stratégies de NGS : identification de variants, séquençage d’éxome, séquençage sur des régions précises…
– Les problématiques en bioinformatiques : stockage et analyse de données, développement de solutions logicielles adaptées…
– Les différentes analyses ainsi que des listes de logiciels pour répondre aux besoins: assemblage denovo et sur génome de référence, annotation et prédiction fonctionnelle, autant open-source que sous licence payante.
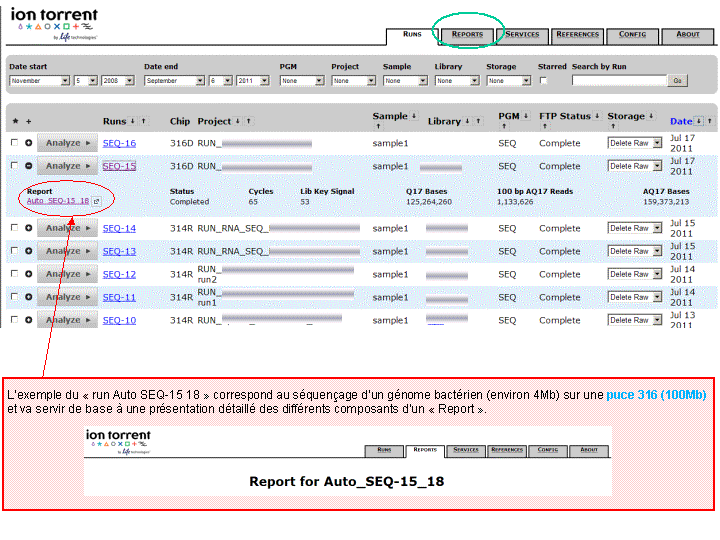
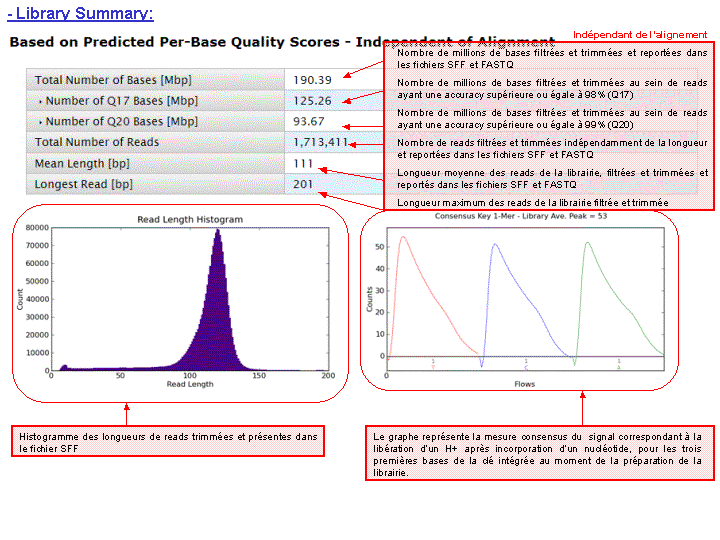
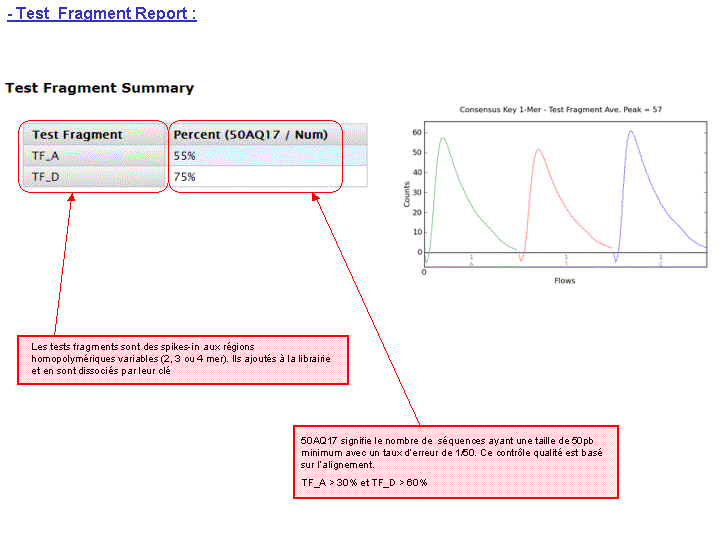
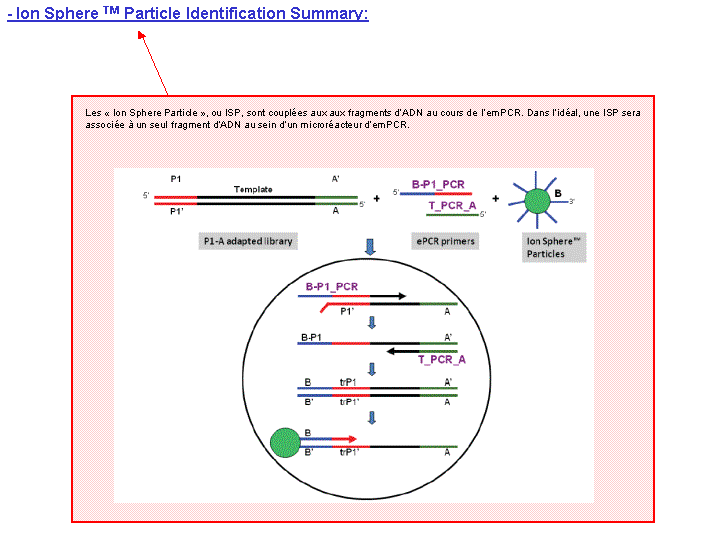
A l’issue d’un « run » de séquençage Ion Torrent (PGM), l’ensemble du signal brut (ionogramme) est converti en séquences et stocké au niveau du serveur. Pour chaque « run de re-séquençage», un alignement préliminaire est réalisé sur la base du génome de référence mentionné lors de l’initialisation du PGM. Cette information est reprise au travers d’un rapport qui comporte également un ensemble de paramètres, que l’on se propose de détailler ci-dessous :
(Cliquer pour agrandir) 
Le rapport se divise en 5 sections:

")

")
")





Depuis 2005, le séquençage haut débit, comme son nom l’indique, a permis d’accroître la quantité de séquences produites par unité de temps, d’individu et de machine. Si intrinsèquement le terme de révolution est associé à ce type de technologies, il semble qu’il serait plus indiqué de l’associer aux nouvelles approches qui en découlent.
En effet, le séquençage nouvelle génération permet d’aborder des études sous de nouveaux angles d’approches. Bien souvent ces approches existaient pour la plupart avant l’avènement de ces nouvelles machines mais leur mise en œuvre étaient bien souvent laborieuses, coûteuses. Beaucoup de techniques nécessitaient des a priori techniques ou scientifiques (des a priori dus à la sélection et aux designs de sondes déposées ou synthétisées sur un support solide dans le cas des puces à ADN permettant les études transcriptomiques). Les nouvelles méthodes de séquençage, quant à elles, permettent de lever certaines anticipations expérimentales. Ainsi une étude du niveau de modulation des transcrits peut grâce à l’emploi de ces technologies en découvrir de nouveaux, ce que ne permet pas ce même type d’études sur puces à ADN. En outre tout a priori constitue un biais expérimental potentiel.
Pour résumer, un peu simplement, le séquençage haut débit dépasse l’outil analytique pour devenir une méthode exploratoire à part entière.
L’objectif de cet article est de proposer un bref aperçu du spectre d’applications et des champs d’expertises que ces nouvelles approches révolutionnent (nous reviendrons plus tard plus en détail sur certaines).
Concernant les applications ayant pour finalité les études génomiques, sont à distinguer :
– Séquençage de novo
Cette application découle de la quantité même de séquences que ces nouvelles générations de machines sont capables de générer. Aujourd’hui il est admis qu’un séquençage de novo nécessite une profondeur de 25 X, c’est-à-dire qu’il est possible de séquencer l’ADN d’un organisme procaryote en un run de séquençage sur la plupart des configurations de séquenceurs. Ce type d’applications a pleinement bénéficié des outils bio-informatiques au niveau des logiciels, machines et compétences humaines de plus en plus disponibles pour tenter de banaliser cette application. Ainsi, le centre de Shenzen avec le BGI (Beijing Genomics Institute, Chine) propose deux projets (pompeusement intitulés library of digital life) le premier consiste au séquençage (et reséquençage) de 1000 génomes de plantes et d’animaux et de 10 000 génomes microbiens.
– Découvertes de SNPs (Single Nucleotide Polymorphisms)
Cette application a très vite trouvé une application directe, elle a contribué au développement de puces à ADN de génotypage haut débit. Ainsi Illumina a pu produire en quelques mois des puces à ADN permettant le génotypage en parallèle d’environ 2,5 millions de SNPs par échantillon (un format 5 millions de SNPs est en préparation) en s’appuyant sur les résultats du consortium 1000 génomes qui a enrichi les bases de données en variations génétiques de la séquence consensus humaine. Ces outils permettant de réaliser des études d’association en réalisant des profils génétiques de plus en plus résolutifs.
Pour ce type d’application, deux modalités sont à distinguer :
– le reséquençage ciblé de zones d’intérêt, étape faisant suite, par exemple, à une étude d’association et permettant après reséquençage d’un locus génétique associé à un caractère particulier, de déterminer la causalité du phénotype différentiellement observé en terme de séquence.
– le réséquençage exhaustif d’ADN génomique. Cette modalité quant à elle, permet la mise en œuvre d’études de comparaisons génétiques de souches (telle que le permet la CGH (Comparative Genomic Hybridization) en s’affranchissant de toute hybridation grâce au séquençage direct)
Sous ce champ expérimental des études génomiques, peuvent être classées toutes les études de métagénomiques où un milieu cherche à être caractérisé le plus exhaustivement et finement possible par la diversité et le degré de contribution de chaque micro-organisme vivant (ou mort…) qui le compose. Le séquençage haut débit permet de rendre accessibles ce type d’approches.
Etudes transcriptomiques
Des méthodologies employées sur puces à ADN telles que les études transcriptomiques ont évolué et été adaptées sur les plateformes de séquençage haut-débit. En outre, ces nouvelles approches permettent de mesurer plus finement des niveaux de modulation tout en tenant compte des isoformes des transcrits. Encore une fois, le fait de séquencer permet de limiter les biais en comparaison de l’emploi de puce à ADN. La lecture plus directe s’affranchit des éléments de design de sondes, des phénomènes d’hybridation etc. A terme, les séquenceurs haut débit supplanteraient les puces à ADN pour ce qui concerne les applications d’études transcriptomiques.
– Réalisation de profils d’expression globale où l’intégralité du transcriptome cherche à être finement caractérisé pour une condition donnée.
– Caractérisation d’ARN non codant
Lors d’études de profils d’expression, les méthodes de séquençage haut débit permettent d’envisager la détermination et caractérisation des ARNs non codants (ici sont particulièrement visés les miRNA et smallRNA).
Etudes épigénétiques
– Etudes de la méthylation de l’ADN (méthyl-seq)
Les études de la méthylation de l’ADN génomique cherche à cartographier les loci fortement métyhylés dans une circonstance donnée. Pour rappel, une faible méthylation favorise la transcription mais une forte méthylation, au contraire, l’inhibe. Lorsque le promoteur d’un gène est méthylé, le gène en aval est réprimé et est donc plus difficilement ou pas du tout transcrit en ARNm.
– Etudes d’association protéines-ADN
Le ChIP-séquençage, également connu sous l’appellation de ChIP-Seq, est utilisé pour analyser des interactions protéines/ADN. Le ChIP-Seq combine immunoprécipitation des zones génétiques sur lesquelles se trouvent fixées des protéines (ChIP) avec le séquençage haut débit de l’ADN afin d’identifier des motifs consensus. Il peut être utilisé pour une cartographie précise de sites de liaison pour une protéine d’intérêt.
Ces deux dernières applications ont dans un premier temps été développées sur la base des tiling-arrays. Le séquençage haut débit permet de diminuer les coûts d’investigation tout en gagnant en sensibilité.
Des technologies émergentes permettent souvent d’envisager de nouvelles applications diagnostiques. Ainsi quelques études depuis 2008, (Fan et al., Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proceedings of the National Science Academy of the USA, 2008, 105, 16266–71), semblent ouvrir la porte à un diagnostic prénatal non invasif.
La recherche médicale avec son débouché clinique de la médecine personnalisée entrevoit des applications au séquençage haut débit. Ainsi, une équipe, montre dans des résultats publiés dans Nature Genetics, pour la troisième fois au monde, la faisabilité d’une étude menée sur la base du séquençage de l’exome, aboutissant à la découverte d’une causalité génétique (simple, puisque monogénique). Cette mutation du gène NOTCH2 causalité d’une maladie rare, le syndrome de Hadju Cheney, une ostéoporose sévère, a été identifiée et caractérisée efficacement par l’application des techniques de séquençage haut débit. Il y a peu ce type d’identifications n’auraient pas été financées puisque trop longue à mener, trop coûteuses pour des retombées certainement perçues comme limitées. Pour beaucoup, ces études menées à grandes vitesses trouveront des applications concrètes dans le champ de la médecine personnalisée… mais de cela nous reparlerons.
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.


{kind=link}
{kind=link}
{kind=link}
{kind=link}