
Currently viewing the category:
"Analyse de données"
Cette article fait suite à notre post sur l’intervention du professeur Arnaud Fontanet de l’Institut Pasteur sur le Coronavirus COVID-19.
Dans sa présentation, le professeur Fontanet renvoie vers trois sites web qui permettent de mieux comprendre le coronavirus.
Chacun dans leur contexte (observation/simulation/étude), ces sites montre la rapidité avec laquelle les chercheurs peuvent développer des outils bioinformatiques de data visualisation pertinents pour la communauté.
Ceci étant bien sur rendu possible à partir du moment où le partage de données épidémiologiques, génétiques, génomiques (…) est effectué.
Pour observer :
Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE
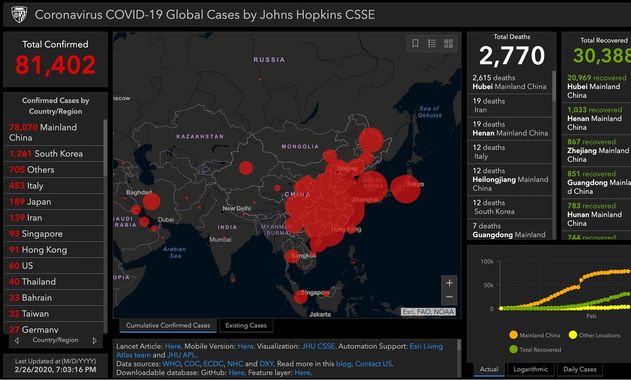
L’université John Hopkins maintient une carte avec des données en temps réel sur le nombre de patients diagnostiqués avec le nouveau coronavirus, le nombre de patients décédés et le nombre de patients guéris. Ces chiffres sont basés sur des informations provenant, entre autres, de l’Organisation mondiale de la santé (OMS) et du Centre européen de prévention et de contrôle des maladies (ECDC). Il peut y avoir de légères différences dans les chiffres réels .
Pour connaître les derniers chiffres confirmés, nous renvoyons aux sites web de l’OMS et de l’ECDC
Github – entrepôt de données : https://github.com/CSSEGISandData/COVID-19
Pour anticiper :
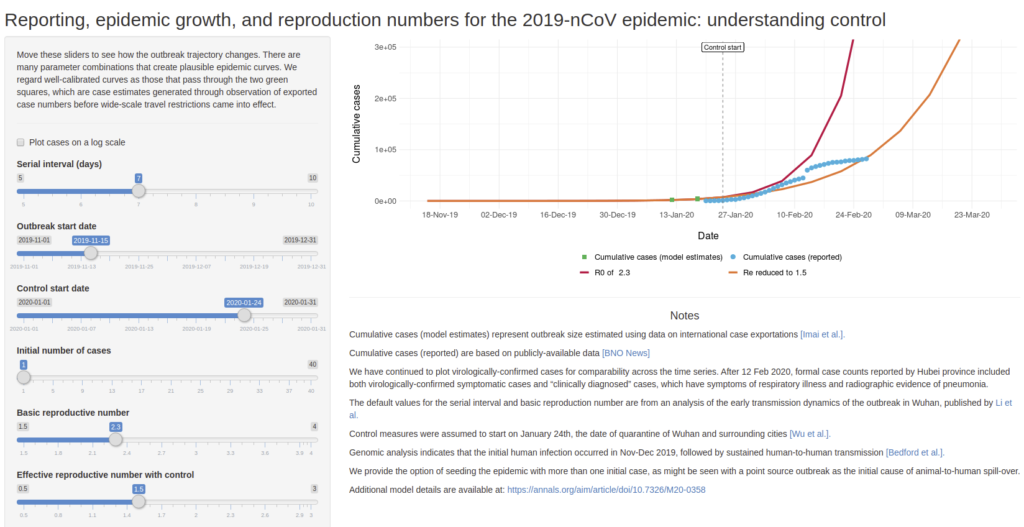
Permet de simuler des scénario de croissance de l’épidémie de COVID-19 en faisant varier quelques paramètres comme :
Serial interval (days) : nombre de jours avant de tomber malade
Outbreak start date : date de début de la maladie
Control start date : date de mise en place de controle (quarantaine, confinement,…)
Initial number of cases : nombre de cas initialement détectés
Basic reproductive number : nombre de personne à leur tour infecté par un malade si aucun contrôle n’est mis en place
Effective reproductive number with control : nombre de personnes à leur tour infecté par un malade si un contrôle est mis en place
Développé par Ashleigh Tuite et David Fisman, Dalla Lana School of Public Health, Université de Toronto
Pour étudier :
Genomic epidemiology of novel coronavirus (HCoV-19)
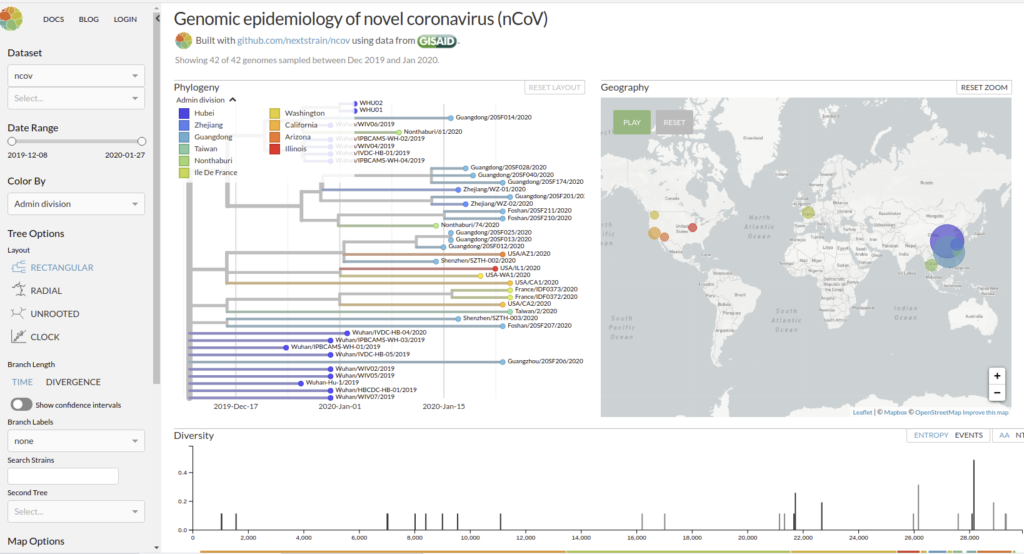
Nextstrain est un projet à open-source visant à exploiter le potentiel scientifique et de santé publique des données sur le génome des agents pathogènes. ils fournissent une vue continuellement mise à jour des données accessibles au public ainsi que de puissants outils d’analyse et de visualisation à l’usage de la communauté. L’objectif est d’aider à la compréhension épidémiologique et d’améliorer la réponse aux épidémies.
Il permet de visualiser les divergences phylogeniques entre les différentes génomes de COVDIR-19 séquencés à ce jour [ 20/02/2020 ]
En savoir plus : Hadfield et al., Nextstrain: real-time tracking of pathogen evolution, Bioinformatics (2018)
Github de l’application : https://github.com/nextstrain/ncov
on en est pas loin avec cet outil publié aujourd’hui dans Genome Biology


Il y a peu, le système MiSeqDx a été la première plate-forme de séquençage haut-débit approuvée par la FDA (US Food and Drug Administration) pour le diagnostic in vitro (IVD). Ceci élargit encore les applications de ces couteaux suisses de la génomique pour aller flirter avec les promesses d’une médecine de précision. C’était, en partie, le souhait des promoteurs des séquenceurs de paillasse (benchtop sequencer) du MiSeq en passant par le Ion Torrent pour aller jusqu’au prometteur séquenceur-clé USB, d’Oxford Nanopore. Ce dernier permet une analyse en temps réel des données générées par le séquenceur. Ce mode opératoire, le temps réel, trouve tout son sens dans le cadre d’applications cliniques où le temps est l’ennemi du clinicien.
Alors que les mappeurs permettant de confronter des reads générés à une référence génomique, sont optimisés pour être de plus en plus rapides, il est très étonnant voire absurde de constater que cette étape ne pouvait être réalisée qu’une fois le run de séquençage, terminé. Aujourd’hui, cet affront fait au bon entendement est en passe d’être réparé dans cette publication, d’octobre 2016, dans Bioinformatics où l’équipe de bioinformatique du Robert Koch Institute propose une première approche dans le sens d’une analyse en temps réel (à base d’extension de k-mers). Une affaire à suivre et un code source disponible : https://gitlab.com/SimonHTausch/HiLive
Source de l’article : HiLive – Real-Time Mapping of Illumina Reads while Sequencing, Bioinformatics. 2016 Oct 29

Le développement des technologies à haut-débit dédiés aux petits ARNs non codant, récemment identifiés (fin des années 90), voit régulièrement déferler des solutions commerciales et libres pour l’analyse gene ontology.
Ce poste est l’occasion de mettre en exergue « miRSystem« , l’un des rares systèmes d’analyses intégrés, gratuit, et intuitif permettant la prédiction de gènes cibles et leurs pathways associés à partir d’une liste de miRs d’intérêt.
La puissance de cet outils réside dans:
1) l’intégration de sept programmes bien connus de prédiction de gènes cibles (DIANA, miRanda, miRBridge, PicTar, PITA, rna22 et TargetScan – cf fig. ci-dessous, rectangles blancs), et qui pour la plupart d’entre eux sont incapables de gérer une analyse englobant plusieurs miRs.
2) l’incorporation de deux algorithmes pour la caractérisation des fonctions biologiques et pathways sur la base de la prédiction des gènes cibles et faisant appel à cinq bases de données (KEGG, Biocarta, PID, Reactome et Gene Ontology – cf fig. ci-dessous, rectangle orange).
Citation
PLoS One. 2012;7(8):e42390. doi: 10.1371/journal.pone.0042390. Epub 2012 Aug 1.
miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets.
Lu TP1, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, Chuang EY.

En ce début d’année, cet article est l’occasion d’aborder rapidement les divers axes de développements, les différents acteurs du séquençage haut-débit de deuxième génération.
– Commençons par Life Technologies et sa gamme Ion Torrent. En fin d’année 2013, la Ion Community (forum où se retrouvent les utilisateurs de la technologie Ion Torrent) s’agite à l’annonce de 3 nouveautés majeures (early access program) :
(i) L’accès à une nouvelle chimie de séquençage, la Hi-Q ™, permettant d’accroître la fiabilité de séquençage. Les erreurs seraient réduites de 90 %, ceci même au niveau des homopolymères, et pour des reads de 400 bases, témoignage de Dag Harmsen à l’appui ! En clair, il semble que ce soit l’enzyme (what else ?) qui ait été remplacé.
(ii) La deuxième annonce concerne l’arrivée de la chimie Avalanche où plusieurs heures d’amplification clonale à l’aide d’un automate One-Touch peuvent être remplacées par l’emploi d’un tube, ce qui prend alors 2 heures pour obtenir une librairie de 500 pb, et ce, de façon isothermique. Un choc de simplification qui ravira les utilisateurs pour lesquels cette étape est limitante.
(iii) La troisième annonce concerne la mise à disposition de kits permettant de réaliser des analyses métagénomiques ciblées 16 S. Un système exploitant le PGM et sa capacité de produire des reads de 400 pb. L’inconnu ici réside dans la mise à disposition de la communauté d’un pipeline analytique performant.
– Qiagen, qui n’est pas connu pour être un acteur de poids sur la scène du séquençage haut-débit, arrive en force en cette année 2014 avec une solution intégrant tous les jalons nécessaires à la complétude d’une étude. Fort de son rachat d’une solution de séquençage (lire l’article : Qiagen investit… le séquençage haut-débit de 2ème génération), Qiagen propose un environnement logiciel des plus intéressants ! En effet, la société néerlandaise a racheté les sociétés CLC Bio et Ingenuity systems. Ces deux sociétés proposent l’une des toutes meilleures solutions d’analyse de séquences: une solution d’assemblage de novo réellement performante grâce à CLC genomics workbench, et Ingenuity systems proposant les pipelines d’analyses suivants: IPA, pour donner un sens biologique aux données omiques, Ireport pour l’analyse de données d’expression et Variant Analysis, un pipeline permettant d’optimiser la recherche de mutations causales.
Ainsi QIAGEN, à l’instar de ce que nous avons tâché de représenter par le schéma ci-dessous, possède actuellement tous les maillons (ou pas loin) d’une chaîne allant de l’échantillon à l’analyse finale traduisant des données de séquences en sens biologique.

– Illumina, quant à elle, semble avoir l’ambition de devenir une sorte de Apple de la « génomicosphère ». En effet, Illumina propose BaseSpace, un Itunes pour les biologistes. D’ailleurs, notons qu’Illumina propose sur Itunes une application : MyGenome, qui propose « d’explorer un véritable génome humain » et d’afficher des rapports sur les variations génétiques importantes. « L’application MyGenome fournit une interface simple, intuitive, et éducative pour vous lancer à la découverte du génome humain« . Revenons à BaseSpace, une interface entre vous et un cloud hébergeant des applications et des données permettant d’analyser les séquences en sortie de MiSeq ou HiSeq. Ce cloud permet aux utilisateurs de délocaliser le stockage de leurs données. L’idée : simplifier au maximum l’analyse par la mise à disposition d’outils et la mise en réseau des utilisateurs. Illumina s’est aperçu que si le goulot d’étranglement constitué par l’analyse de données de séquençage haut-débit volait en éclat, nécessairement les runs pourraient se multiplier avec leur chiffre d’affaire. Le schéma ci-dessous reprend quelques éléments de la solution analytique développée par Illumina.

Une communauté de plus de 12000 utilisateurs, un espace permettant l’utilisation d’une vingtaine d’applications. L’objectif d’Illumina : créer un espace attractif, émulant et incitant les intervenants à mettre à disposition les applications développées en priorité sur cet espace. Anticipant une demande exponentielle d’analyses et d’espace de stockage lorsque le HiSeq a été intégré au BaseSpace, Illumina a décidé de mettre en place une politique de tarification qui limiterait la quantité d’espace libre pour stocker et traiter les données génomiques dans le cloud. En vertu de cette logique, les utilisateurs reçoivent un téraoctet gratuit d’espace pour le stockage et le traitement des données et seraient alors en mesure d’acheter du stockage supplémentaire par incréments de téraoctet ou 10 téraoctets – un téraoctet coûterait 250 $ par mois ou $ 2,000 d’avance pour une année complète , tandis que 10 téraoctets seraient à 1500 $ par mois ou une avance des frais annuels de $ 12 000 (données chiffrées début 2013).
En conclusion, si les années précédentes ont vu le lancement de nouveaux séquenceurs, avec depuis 2011 l’arrivée de séquenceurs de paillasse, les années 2013-2014 attendent la diffusion de séquenceurs de 3ème génération. Qiagen est un petit nouveau dans la course, ce nouvel acteur est capable, sans réel développement, de proposer une solution complète grâce à une stratégie de rachat pertinente. Illumina et Life Technologies, pendant ce temps, poursuivent leur développement en essayant d’émuler les utilisateurs avec, respectivement, leur BaseSpace et Ion Community. L’opérateur historique, Roche est le grand silencieux avec une stratégie peu lisible…

Le séquençage haut-débit voit cohabiter depuis quelques années deux générations de séquenceurs.
Au passage, une question Trivial Pursuit pour laquelle il faudra avoir un œil de caracal : quelqu’un sait quelle société a développé la première génération de séquenceurs haut-débit ? et quand ?
Les séquenceurs de deuxième génération se voient conditionnés sous forme de séquenceurs de paillasse (PGM de Ion Torrent, Miseq d’Illumina, GS-junior de Roche) permettant une démocratisation du séquençage, pendant que leurs grands frères pulvérisent la loi de Moore pour envisager un rendement (coût / Mb) toujours plus compétitif.
La large diffusion du séquençage de 3ème génération se laisse désirer laissant le champ libre à la génération précédente. Cet article vise à réaliser un court état des lieux du séquençage haut-débit de troisième génération : un futur plus ou moins lointain, de nouvelles applications potentielles.
La question : séquenceurs de 3ème génération, l’âge de raison, c’est pour quand ? est l’interrogation qui a hanté l’AGBT 2013 marqué par le silence d’Oxford Nanopore. Cette année 2013 fut marquée par le retrait d’Illumina du capital de la société britannique : « Oxford Nanopore Technologies Ltd a annoncé la vente d’une participation détenue par son concurrent américain Illumina Inc., une étape vers la fin d’une relation pleine de conflits dans la course au développement des séquenceurs haut-débit permettant de séquencer plus rapidement et pour moins cher. »
Avant de caractériser ce que sont, seront, pourront être les 3ème générations de séquenceurs, commençons par un rapide tour des caractéristiques générales de leurs prédécesseurs et principalement de ce qui constitue leurs points faibles :
– la phase d’amplification clonale (réalisée par PCR) est source de biais (doublons, erreurs de PCR)
– les problèmes liés au déphasage engendrant une chute de la qualité le long du read produit (ce qui bride la production de reads vraiment longs)
-des reads courts (de moins d’une centaine à environ 800 bases – vous l’aurez noté ce point est en partie une conséquence du précédent)
-des machines et des consommables onéreux
– des temps de run longs
Ainsi l’objectif principal des séquenceurs de 3ème génération est de palier les défauts de leurs aînés en produisant des reads plus longs, plus vite pour moins cher. Les séquenceurs de 2ème génération, quels que soient leurs modes de détection (mesure de fluorescence, mesure de pH) sont trop peu sensibles pour envisager la détection d’une simple molécule, d’un simple nucléotide : nécessairement la librairie doit être amplifiée, ce qui provoque des biais, des temps de préparation relativement longs et l’usage de consommables qui impacte le coût final de séquençage… assez rapidement la qualité chute plus vos reads s’allongent ce qui oblige à brider les tailles de reads que ces technologies sont capables de délivrer. En outre, travaillant sur une matrice qui est une copie de votre librairie initiale, l’information portée par les bases méthylées est perdue (ceci oblige à ajouter une phase de traitement au bisulfite qui peut être hasardeuse)
Actuellement l’une des seules technologies de 3ème génération réellement utilisée est celle de Pacific Biosciences (les hipsters disent « PacBio »). La firme, fondée en 2004, a lancé en 2010, son premier séquenceur de troisième génération le Pacbio RS basé sur une technique de séquençage SMRT (Single Molecule Real Time sequencing.) Aujourd’hui la société Roche qui n’a pu absorber Illumina lors de son OPA, a investi 75 millions de USD, le 25 septembre 2013, pour co-développer des kits diagnostiques in vitro exploitant la technologie de PacBio.
La technologie de PacBio est aujourd’hui exploitée pour réaliser du séquençage de novo de petits génomes :

Avec ces 200 à 300 Mb délivrés par SMRT-cell, séquencer des organismes eucaryotes supérieurs demande un investissement important, malgré tout, cette technologie délivrant des reads de plusieurs milliers de bases, permet d’envisager une diminution du nombre de contigs obtenus par les seules stratégies reads-courts / gros débit.
Face à la technologie proposée par PacBio, d’autres technologies essaient d’émerger pour arriver à occuper le marché du séquençage de 3ème génération :
– La combinaison détection optique et multipore est une voie envisagée pour le séquençage de 3ème génération avec le travail mené par NobleGen biosciences.
– L’imagerie directe de l’ADN
Le microscope électronique offre une résolution possible jusqu’à 100 pm, de sorte que les biomolécules et les structures microscopiques tels que des virus, des ribosomes, des protéines, des lipides, des petites molécules et des atomes même simples peuvent être observés. Bien que l’ADN est visible lorsqu’on l’observe avec un microscope électronique, la résolution de l’image obtenue n’est pas suffisamment élevée pour permettre le déchiffrement de la séquence, c’est à dire, le séquençage de l’ADN. Cependant, lors du marquage différentiel des bases de l’ADN avec des atomes lourds ou des métaux, un tel séquençage devient possible.

– Le séquençage à l’aide de transistor (Transistor-mediated DNA sequencing– une technologie développée par IBM)

Dans le système conceptualisé par IBM, l’ADN est contraint de passer par le pore à cause de la tension électrique subie, la vitesse de passage de la molécule à séquencer est maîtrisée à l’aide de contacts métalliques à l’intérieur du nanopore. La lecture des bases serait réalisée lors du passage de l’ADN simple brin au travers du pore (ça rappelle quelque chose…)
– Et Oxford Nanopore dans tout cela ? Si la société anglaise a annoncé la vente de la participation d’Illumina, elle a marqué l’année 2013 par son silence assourdissant. Passé l’oxymore, en cette fin d’année, coup de poker ou réel lancement, Oxford Nanopore propose un programme d’accès à sa technologie Minion où pour 1000 USD, il est possible de postuler à l’achat des clés USB de séquençage.
La stratégie d’Oxford Nanopore est basée, en partie, sur la possible démocratisation du séquençage de 3ème génération, elle s’oppose à celle de PacBio qui mise sur son arrivée précoce sur le secteur du séquençage haut-débit : décentralisation contre l’inverse. En clair, l’investissement d’un PacBio est tel que l’outil est réservé à des centres, des prestataires de services pouvant assumer cet investissement, ce qui oblige à centraliser les échantillons pour les séquencer, contre les produits (encore en développement) d’Oxford Nanopore dont la promesse est : le séquençage pour tous (ou presque).
PacBio revendique sa participation à un projet qui consiste à doubler la quantité de génomes bactériens « terminés » (actuellement de 2384) en quelques mois.

En cliquant ci-dessus sur la représentation graphique qui illustre la différence de plasticité de génome entre le génome d’une Bordetella pertussis (l’agent pathogène responsable de la coqueluche) et celui d’Escherichia coli, un poster vous apparaîtra. Ce dernier reprend les caractéristiques de l’utilisation de la technologie de PacBio à des fins d’assemblage de novo de génomes bactériens par une stratégie non-hybride (seuls des reads de PacBio sont utilisés). Les résultats sont assez bluffants, la longueur des reads de PacBio permet un assemblage complet (au prix de plusieurs SMRT cells tout de même !), de génomes bactériens « difficiles » tel que celui de Bordetella pertussis connu pour posséder un GC % relativement élevé (environ 65 %) ainsi que de nombreux éléments transposables. Les génomes possédant de nombreux éléments répétés posent de grandes difficultés d’assemblage, c’est un des arguments qui permet à PacBio de positionner sa technologie actuellement… en quelques mois les stratégies hybrides (reads courts générés par des séquenceurs de 2ème génération) ont laissé place aux stratégies non-hybrides où le séquençage PacBio se suffit à lui-même.
La diversité du parc technologique des séquenceurs de deuxième génération n’est plus une surprise pour personne. Ceci étant, il devenait indispensable de mettre à jour l’ensemble des informations postées sur ce site, il y a exactement deux ans (2011), faisant un état de l’art des différentes caractéristiques technologiques des séquenceurs, ainsi que les possibles applications biologiques associées.
Pour ne mentionner que les trois plus gros fournisseurs du marché, les sociétés Roche, Illumina et Life Technologies n’ont cessé de faire évoluer leur gamme, tant sur le plan des équipements que sur le plan des capacités de séquençage.

Par voie de conséquence, ce survol est l’occasion de refaire le point sur les technologies appropriées selon l’application biologique recherchée. A noter que le Ion proton, dernier en date sur le marché des séquenceurs de deuxième génération disposera au cours de l’année 2014 d’une puce « PIII » permettant de générer environ 64Gb. Cette capacité de séquençage permettra à Ion torrent de se positionner sur le séquençage de génome humain à partir d’un séquenceur de paillasse et accèdera ainsi à la gamme complète des applications citées ci-dessous.

 L’accélération du débit bibliographique faisant référence aux « miRNA » atteste aisément de leur caractérisation récente (Lee RC et al., Cell (1993)) et de l’intérêt lié à leurs potentielles fonctions .
L’accélération du débit bibliographique faisant référence aux « miRNA » atteste aisément de leur caractérisation récente (Lee RC et al., Cell (1993)) et de l’intérêt lié à leurs potentielles fonctions .
Il aura fallu près de dix années supplémentaires pour mettre en évidence leur implication en tant que régulateurs biologiques (notamment au niveau de la régulation de l’expression des gènes) et leurs impacts dans de certains cancers… Aussi, le développement des nouvelles technologies de séquençage à haut débit contribue forcément à cette émergence.
Ce poste est l’occasion de présenter « miRNAtools » qui comme son nom l’indique, regroupe un grand nombre de liens renvoyant vers différents outils dédiés aux miRNA.

– Analyse de données NGS appliquées aux miRNAs (étude des profils d’expression). La liste des 7 softwares présentés n’est pas exhaustive et en voici quelques uns supplémentaires à tester: « mireap », « miRTRAP », « DSAP », « mirena », « miRNAkey », « SeqBuster », « E-mir », … . Une comparaison de l’efficacité de certains de ces outils fera l’objet d’un prochain poste.
– Prédiction de cibles selon les miRNA étudiés.
– Analyse de pathways impliquant les miR d’intérêt. Pour cette dernière application, le soft DIANA LAB – Mirpath proposé, bien que facile d’utilisation et gratuit, a le défaut de ne s’appliquer qu’aux organismes « humain » et « souris ». Dans ce registre et moyennant quelques milliers d ‘euros, « Ingenuity Pathway Analysis » (« IPA ») reste de loin l’outil idéal. En effet, en plus d’identifier les voies métaboliques au sein desquels sont impliqués les miR modulés comme proposé par Mirpath, « IPA » permet également d’intégrer les résultats de modulation de miR et d’expression de gènes pour une même condition d’étude…
 A l’heure où Jonathan Rohtberg et Craig Venter cherchent de l’ADN sur Mars (lire : Searching for Alien Genomes), l’ADN sur Terre ne serait-il pas à la veille de changer de statut ? En effet, si l’ADN est « le support de l’information génétique »… est-il possible que cette macromolécule devienne le support de l’information tout court ?
A l’heure où Jonathan Rohtberg et Craig Venter cherchent de l’ADN sur Mars (lire : Searching for Alien Genomes), l’ADN sur Terre ne serait-il pas à la veille de changer de statut ? En effet, si l’ADN est « le support de l’information génétique »… est-il possible que cette macromolécule devienne le support de l’information tout court ?
– C’est humblement, dans le domaine de la traçabilité, que l’ADN a trouvé un rôle d’espion : Ainsi, au début des années 2000, la société norvégienne ChemTag, tente de développer des systèmes permettant de tracer dans nos océans, le pétrole retrouvé suite à d’éventuels dégazages. L’idée est simple : l’ADN est utilisé comme un code-barre, la succession du code de quatre lettres permet assez rapidement d’obtenir un code (4^n) ne permettant qu’avec une probabilité infime d’être retrouvé par hasard. Il faut savoir que par année, plus d’un million de tonnes de pétrole sont déversées dans les océans. La problématique en vaut la peine, Poséïdon en sera gré. Ici toute la difficulté qui se présentait à la société ChemTag : faire en sorte que l’ADN ait une grande affinité pour le pétrole (évidemment en cas de dégazage si le traceur ADN se dissout dans l’océan… adieu code-barre) et permettre que ce traceur soit purifié le plus simplement possible. L’ADN est une molécule plutôt stable et plutôt facile à « lire », ne modifiant pas les qualités organoleptiques d’une substance… et surtout cette molécule permet un nombre infini de combinaisons.
Moult industriels souhaiteraient être capables d’identifier en toute objectivité (légalement) tout organisme vivant leur appartenant (une souche de production par exemple). A cette fin, le code barre biologique peut être utilisé de deux manières :
– artificiellement, en « incorporant » par manipulation génétique une séquence connue. Cette option est bien souvent écartée parce qu’elle fait appel à la notion d’OGM, mention qui peut effrayer le consommateur.
– naturellement, en connaissant le patrimoine génétique du génome employé dans un procédé industriel. Cela implique de déterminer une séquence singulière qui n’appartiendrait qu’à cette organisme. Souvent ce type de procédures fait appel à une combinaison de séquences ou de loci polymorphes.
L’évolution des techniques de séquençages (augmentation des débits, diminutions des coûts, rapidités des runs) a permis de rendre accessible une séquence complète d’un micro-organisme ou une carte de SNPs haute-densité pour certains eucaryotes supérieurs dont on dispose de puces SNPs.
Aujourd’hui, certains envisagent d’exploiter l’ADN pour en faire un support robuste de l’information numérique.

Depuis quelques semaines, une petite bataille a lieu entre l’équipe de Georges Church de la Medical School de Boston et Christophe Dessimoz de l’EBI.
Le premier a encodé un livre dont il est le co-auteur : Regenesis: How Synthetic Biology Will Reinvent Nature and Ourselves in DNA comportant 53000 mots et 11 images en jpeg accompagné d’un programme Javascript pour un total de 5,37 Mo dans un picogramme d’ADN (un peu moins de 1 Gbase). Le second a encodé du Shakespeare, un discours de Martin Luther King, une photo et la copie d’un article de 1953 décrivant la structure de l’ADN… on s’amuse plutôt pas mal aussi de ce côté de l’Atlantique !
Ces querelles d’encodeurs ont malgré tout un intérêt. Ils démontrent la faisabilité de ce type de système et montrent qu’il est désormais envisageable de sauvegarder pour des échelles de temps très longues, de grandes quantités d’informations qui échapperaient à bien des autodafés.
Les systèmes d’encodage au sein même de l’ADN relégueront nos séquenceurs haut-débit au niveau de lecteurs DVD ou Blu Ray du futur… La limite du système, actuellement, tient plus des synthétiseurs d’ADN et des technologies de séquençage qui n’ont pas réellement été conçus à cette fin. Mais à l’heure où l’on promet d’ici quelques dizaines de mois un séquençage de génome humain pour une centaine de dollars contre quelques milliards il y a 10 ans, tous les espoirs sont permis pour que l’information séculaire contenue dans une bibliothèque patrimoine de l’Humanité devienne une bibliothèque d’Alexandrie du futur, franchissant les âges, échappant aux catastrophes naturelles, aux censures et à l’oubli.
 Lors d’articles précédents nous vous avions présenté le logiciel de Workflow Galaxy, qui permet d’analyser et de visualiser toutes sortes de données biologiques à partir d’une interface simple d’utilisation.
Lors d’articles précédents nous vous avions présenté le logiciel de Workflow Galaxy, qui permet d’analyser et de visualiser toutes sortes de données biologiques à partir d’une interface simple d’utilisation.
Galaxy est en fait une brique d’une collection d’outils dédiés à l’analyse et au stockage de données biologiques : GMOD ( Generic Model Organism Database )
Le lundi 14 mai a été l’occasion pour nous d’assister à une conférence sur l’utilisation de certains des outils GMOD, dont voici les principaux enseignements :
> Le projet GMOD a pour objectif de fournir à l’utilisateur biologiste un ensemble d’outils interconnectés, libre de droit (open-source), générique (pour tous types de données biologiques) et facile d’utilisation (à travers des services Web principalement)
> Les outils GMOD sont développés (et donc installés) par des bioinformaticiens pour une utilisation par des biologistes.
> Certains outils sont indispensables à GMOD, pour la manipulation de données génomiques, c’est le cas des outils Chado et Gbrowse qui sont respectivement les squelettes pour la manipulation et pour la visualisation des données biologiques.
Le schéma ci-dessous décrit les modules et interactions présentés lors de cette journée thématique :

GMOD - Modules et interactions présentés le 14 juin
En résumé :
> Chado est un schéma générique de base de données relationnelles pour le stockage de tous types de données biologiques.
Description détaillée : GMOD-CHADO
> GBrowse est un outil de visualisation de données biologiques très puissant et certainement l’outil le plus populaire de la suite GMOD
Description détaillée : GMOD-GBROWSE
> Biomart est un outil de recherches avancées (ou requêtes complexes) pour la base de donnée relationnelle Chado
> Apollo est un module pour la correction manuelle d’annotation structurelle
Description détaillée : GMOD-APOLLO
> Tripal est une interface web développée en PHP pour interrogation de Chado
Pour aller plus loin : Détails techniques sur l’utilisation des outils GMOD :

Environnement informatique : CHADO fonctionne par défaut avec Postgres-Sql. Pour l'interfaçage avec Tripal, la solution étant développée avec PHP, il est nécessaire d'installer un serveur Web Apache. Gbrowse peut s'utiliser à partir de l’interface Tripal mais également en "stand-alone". Afin d'accélérer la visualisation des données il est vivement recommandé d'utiliser les adapteurs Bio::DB::*, soit en relais entre Chado et Gbrowse (présentés dans cet article), soit en dupliquant les informations dans les deux bases de données (la visualisation ne se connecte qu'à Bio::DB sans passer par CHADO). Intuitivement, nous privilégions la première solution qui n’entraîne pas de duplication.
Pour conclure, GMOD propose un ensemble de modules pour la standardisation des processus bioinformatique : stockage et manipulation de données biologiques, visualisation et analyses avancées (assemblage, annotation…).
L’utilisation de tels outils (open-source) va dans le bon sens pour le partage scientifique et la standardisation des processus utilisés lors de l’analyse bioinformatique. Cela n’était pas le but de la conférence à laquelle nous avons assistée mais il serait également intéressant de connaître les conditions pour intégrer ces propres outils bioinformatiques en tant que brique GMOD.
GMOD est donc intéressant si :
> vous souhaitez stocker et gérer vos données biologiques
> vous cherchez des solutions d’analyses bioinformatiques déjà développées et robustes
Les biologistes sont les utilisateurs des outils GMOD, en revanche l’installation, l’administration et la formation des utilisateurs ne peuvent échapper à l’intervention, au moins ponctuelle, d’un bioinformaticien.
Si vous projetez d’utiliser ces outils, nous vous conseillons donc dans un premier temps, de regrouper l’ensemble des acteurs, installateurs comme utilisateurs de GMOD, afin de présenter les solutions offertes par l’outil et déterminer les besoins et objectifs pour votre propre utilisation.
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.

