
Currently viewing the category:
"Diagnostic"
Des explications précises et instructives sur l’état actuel (au 20 février 2020) des connaissances concernant l’épidémie provoquée par ce nouveau Coronavirus (COVID-19) – Durée : 35Min
Le professeur Arnaud Fontanet revient en détail sur l’histoire de ce virus, de la découverte des premiers cas, à l’enquête sur son mode de transmission jusqu’à son séquençage extrêmement rapide.
Il donne également beaucoup d’informations sur la durée d’incubation, la contagion, les symptômes associés, les mesures sanitaires et le travail des épidémiologistes et des chercheurs pour endiguer la propagation.
Les conséquences économiques ainsi qu’un parallèle pas inintéressant avec la grippe saisonnière permettront, pour certains, de mettre en perspective cette épidémie par rapport à notre monde actuel…
Cette intervention fait partie du MOOC de l’Institut Pasteur « Virus émergents et réémergents ».
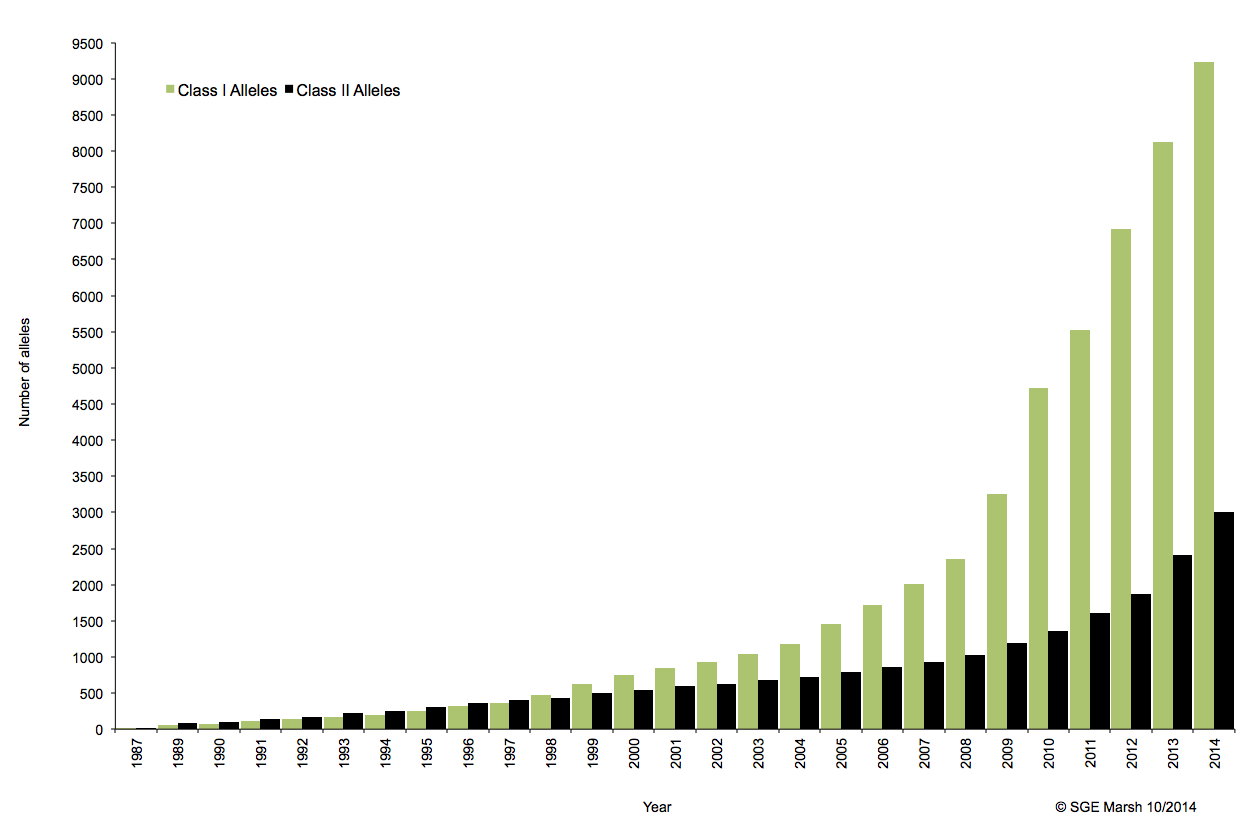
En cancérologie, l’allogreffe de moelle osseuse s’inscrit dans un parcours thérapeutique notamment comme traitement de consolidation après une chimiothérapie. Aussitôt, les notions de rejet ou d’acceptation du greffon apparaissent et il devient indispensable que les systèmes HLA (Human Leucocyt Antigens, découvert en 1950) du donneur et du receveur soient les plus proches possibles.
Ce système immunogène, situé sur le bras court du chromosome 6 chez l’homme, est caractérisé par son polygénisme et son polymorphisme qui sont à l’origine d’une grande variabilité interindividuelle et en fait le déterminant principal du résultat de greffe. L’ensemble des gènes HLA sont subdivisés en trois régions du chromosome 6 qui contiennent chacune de nombreux gènes avec ou sans fonction immunologique. On distingue ainsi la région CMH de classe I, de classe II, et de classe III.
A ce jour, un rendu de typage est ciblé sur une portion génomique restreinte codant pour le HLA. Il s’agit de l’exon 2 et 3 des loci HLA-A, HLA-B et HLA-C (région I), l’exon 2 et 3 des loci HLA-DQ (DQ-A et DQ-B) et l’exon 2 pour HLA-DR (DRA et DRB1), où repose prés de 70% du polymorphisme. La région III ne renfermant pas de gènes intervenant dans la présentation antigénique.

C’est ainsi que différentes approches de typages ont été développées et l’avènement de la technique de PCR au milieu des années 1980 a pallié aux limites de résolution de la sérologie employée jusqu’alors. D’un typage rendu au niveau générique (2 Digits), la « PCR-SSO » (Sequence Specific Oligonucleotide) et la « PCR-SSP » (Sequence Specific Primer) développées dans les années 1990 ont permis d’accéder à un résultat allélique (4 Digits). Cette avancée technologique s’est poursuivie la décennie suivante avec la « PCR-SBT » (Sequence Based Typing) ou séquençage « Sanger » puis plus récemment avec la PCR en temps réel (ex: linkage). Toutes ces techniques de biologie moléculaire ont permis de mettre en avant le polymorphisme et la grande diversité génétique du HLA. Chaque année, de nombreux allèles sont découverts, alimentant continuellement la banque de données de référence IMGT.

Associé à cette augmentation constante du nombre d’allèles typés, le nombre d’ambigüités croît et met progressivement en difficulté les technologies conventionnelles qui atteignent leurs limites. De plus, le pourcentage de réussite des allogreffes n’atteint environ que 50%. L’exploitation du reste de l’information génomique permettrait potentiellement d’améliorer cette performance par un typage plus affiné.
Le recours au séquençage nouvelle génération apparait donc inévitable. En plus de gérer les ambiguïtés par un séquençage allélique, le NGS permet de traiter simultanément de grandes quantités d’échantillons réduisant ainsi le coût unitaire, d’accéder à un niveau de résolution supérieur (4,6 ou 8 digits) tout en ayant la capacité de cibler des loci entier (Long Range PCR).
Ainsi, plusieurs stratégies existent avec leur solution technique adaptée à la préparation de la matrice d’ADN à séquencer:
– L’amplification ciblée des régions d’intérêt par PCR de fusion. Cette approche permets un gain de temps et une réduction des coûts en s’affranchissant des étapes de fragmentation, ligation, et autres purifications… .Par ailleurs elle n’est pas la mieux adapté dans le cas d’une couverture de séquençage de l’ensemble de la région génomique HLA.
– L’amplification par « Long Range PCR » permet une couverture complète des différents loci étudiés. Les fragments de plusieurs Kb subissent alors une fragmentation, une ligation des adaptateurs et indexation. Cette approche permet d’accéder à davantage d’informations (régions exoniques et introniques).
– La capture de séquences par hybridation. Même si cette solution est bien caractérisée elle n’est pas si efficace en terme de capture avec une disparité selon la taille des fragments.
– Le séquençage de génome entier ou d’exome. Cette approche est la moins biaisée et couvre tous les gènes du système HLA. Paradoxalement, l’analyse nécessite beaucoup trop de ressources pour une utilisation en routine et ne permets pas de traiter autant d’échantillons simultanément.
Plusieurs sociétés commerciales proposent des solutions clé en main depuis la préparation des échantillons (avec l’option « Long Range PCR » qui semble la plus plébiscité) jusqu’à l’analyse de résultats via leur logiciel dédié. Certaines sont en cours de validation de méthode pendant que d’autres tentent d’inonder le marché. Parmi elles, Gendx, Omixon, Illumina, One lambda, Life technologies, Immucor, etc…

L’exploitation des capacités et caractéristiques des solutions de séquençage à haut-débit permettrait d’affiner considérablement le typage HLA. Ainsi le décryptage de l’ensemble des régions codantes et non-codantes du génome d’intérêt représente un enjeu important dans la réussite des greffes. Par ailleurs, cette approche nécessitera une mise à jour considérable des banques de données (IMGT) avec une validation de nombreux nouveaux allèles.
La PCR en point final, celle qui se termine souvent par un dépôt sur gel d’agarose, ainsi que la PCR quantitative, dans laquelle on suit l’évolution de la libération de molécules fluorescentes dont le décollage plus ou moins précoce dépend de la quantité de matrice initialement introduite, se voient déclinées en versions numériques. Par numérique est entendu ici, qu’une PCR peut prendre deux valeurs : 0 ou 1 (idéalement, 0 quand aucune cible n’est introduite au départ d’une PCR et que par voie de conséquence aucun signal issu d’amplification n’est observé, et 1 pour l’exact inverse).
Le principe de la PCR numérique est simple au niveau du concept mais techniquement beaucoup plus difficile à mettre en application. Il s’agit de multiplier le nombre de bio-réacteurs disponibles, ainsi, un échantillon sera partitionné en des milliers de compartiments distincts. Ensuite chaque compartiment sera considéré comme autant de réacteurs indépendants… donc en fin de PCR autant de réactions montrant oui ou non une amplification (autant de 0 et de 1), renvoyant à un système binaire. L’application de la Loi de Poisson permettra ensuite d’estimer très finement la quantité initiale de cibles présentes dans l’échantillon de départ.

principe général de la PCR numérique
Les avantages de la PCR numérique par rapport à la qPCR classique sont assez évidents :
– elle permet une quantification absolue sans établir une courbe standard préalable
– elle est plus sensible voire beaucoup plus sensible. Raindance annonce ainsi détecter un mutant parmi 250 000 molécules sauvages avec une limite inférieure de détection de 1 parmi 1.000.000
– elle est moins sensible aux inhibiteurs. Le fait de diluer la matrice ADN complexe de départ pour la répartir dans un nombre important de réacteurs permet de favoriser le départ de PCR
– elle est nettement plus précise. La technologie sera d’autant plus précise que le nombre de compartiments, le nombre de micro-réacteurs sera important
L’article « Digital PCR hits its stride » (La PCR numérique franchit un nouveau cap) de Monya Baker dans le Nature Methods (juin 2012) permet un aperçu de la technologie qui est passée du concept à des solutions commerciales qui se veulent de plus en plus accessibles.
Actuellement, plusieurs fournisseurs proposent des plateformes aux spécifications très différentes. Les quatre fournisseurs ci-dessous proposent des systèmes de PCR numérique basés sur de la microfluidique pour Fluidigm, des interfaces solides (type OpenArray) de Life Technologies et des micro-gouttes pour Raindance et Biorad.

Ainsi, que le mentionne Jim Huggett (chef d’équipe au LGC), cité dans la publication de Monya Baker (Digital PCR hits its stride), la PCR numérique est réservée à des utilisateurs experts et se trouve encore allouée à des applications spécialisées car encore beaucoup plus chère que la PCR quantitative (traditionnelle) qui reste adaptée à la majorité des applications.
 23andMe, comme Knome et Decode, est une société californienne de biotechnologie pionnière dans la génomique personnelle. La société a été lancée en 2006 par Anne Wojcicki, Linda Avey et Paul Cusenza sur la base d’un concept marketing simple et audacieux : mettre des tests génétiques à disposition du grand public en les vendant en direct sur une plateforme internet. Aujourd’hui la PME revendique 45 employés, 20 millions de USD et une omniprésence sur le web. Le modèle de développement de 23andMe passe par une exploitation judicieuse d’un site web, leur meilleur VRP. La photo du couple ci-dessus représente Anne Wojcicki, la co-fondatrice de 23andMe et Sergey Brin le co-fondateur avec Larry Page de Google, la société maîtresse du web. Google a d’ailleurs investi près de 4 millions de dollars dans la PME de Madame dès ses débuts en mai 2007. Notez qu’à l’époque le test génétique vendu par la toute jeune société de biotechnologie coûtait 999 USD (le prix aura donc été divisé par 10 en 5 ans). Pour la petite histoire, la mère de Sergey Brin est atteinte de la maladie de Parkinson, ce qui a incité le milliardaire à réaliser un test génétique en 2006, pour découvrir qu’il était porteur de la mutation du gène LRRK2. Ceci explique son intérêt pour les tests génomiques personnels.
23andMe, comme Knome et Decode, est une société californienne de biotechnologie pionnière dans la génomique personnelle. La société a été lancée en 2006 par Anne Wojcicki, Linda Avey et Paul Cusenza sur la base d’un concept marketing simple et audacieux : mettre des tests génétiques à disposition du grand public en les vendant en direct sur une plateforme internet. Aujourd’hui la PME revendique 45 employés, 20 millions de USD et une omniprésence sur le web. Le modèle de développement de 23andMe passe par une exploitation judicieuse d’un site web, leur meilleur VRP. La photo du couple ci-dessus représente Anne Wojcicki, la co-fondatrice de 23andMe et Sergey Brin le co-fondateur avec Larry Page de Google, la société maîtresse du web. Google a d’ailleurs investi près de 4 millions de dollars dans la PME de Madame dès ses débuts en mai 2007. Notez qu’à l’époque le test génétique vendu par la toute jeune société de biotechnologie coûtait 999 USD (le prix aura donc été divisé par 10 en 5 ans). Pour la petite histoire, la mère de Sergey Brin est atteinte de la maladie de Parkinson, ce qui a incité le milliardaire à réaliser un test génétique en 2006, pour découvrir qu’il était porteur de la mutation du gène LRRK2. Ceci explique son intérêt pour les tests génomiques personnels.
Début 2013, le produit coûte : 99 USD auxquels il faut ajouter 79,95 USD pour les frais de port (ces frais limitent l’accès au vieux continent à 23andMe), la note se monte donc pour un Français à 137,70 € (mi 2011, le prix était de 200 €). Il est facile dorénavant de comprendre que, les volumes des puces Illumina grimpant, la part du coût allouée aux consommables a baissé, mais que la distribution web a ses limites… la poste.
Tests génomiques ?
Le test génétique que propose 23andMe permet :
– d’estimer les facteurs de risques d’être, un jour, atteint d’une maladie à composante majoritairement génétique
– d’estimer vos aptitudes physiques et intellectuelles (en présupposant que la variabilité génétique de ces traits contribue fortement auxdits traits estimés)

– de déterminer son origine géographique, ainsi que, pour le folklore, de quel personnage célèbre dont 23andMe possède le génotype, vous vous rapprochez le plus (sur le ce point lire notre article : A la recherche du chromosome perdu : aux sources des hominidés)
– enfin, 23andMe accumule des génotypages venant alimenter une base de données associée à une population de référence et ainsi accroître l’information génomique disponible pour d’éventuelles recherches (à mener par d’autres…), tout cela financé par les clients de 23andMe… il fallait y penser et oser, c’est chose faite !
Principe ?
Contrairement à un amalgame souvent rencontré, la société californienne n’utilise que très peu le séquençage pour son produit phare. Le principe réside dans l’extraction de l’ADN contenu dans les cellules de la salive, ADN qui fera l’objet d’un génotypage haut-débit permettant de cartographier le génome du client. 23andMe ne fait que compiler les données de GWAS (Genome Wide Association Studies), les études d’association publiées et agglomérées plus ou moins subtilement. Ce type d’études consiste en l’association de profils génétiques à des phénotypes caractérisés. En outre, 23andMe exploite la connaissance des marqueurs en Déséquilibre de Liaison avec une maladie ou une aptitude, le tout en concaténant l’information scientifique publique.
Des témoignages de clients pour en convaincre de nouveaux
Depuis le début 2013, plusieurs témoignages ont été utilisés par la presse pour décrire ce que proposait 23andMe, une publicité à moindre frais. Celui de Slate relate l’histoire de Jill Uchiyama, une personne qui a été adoptée à la naissance et qui à l’âge de 41 ans par l’intermédiaire des services proposés par 23ansMe a retrouvé quelques cousins plus ou moins proches (300 cousins) et en a rencontré un… un manière de recréer de simili-liens de sang. Un autre présent sur le site Quora.com témoigne du cas d’un père découvrant qu’il avait des risques de développer une intolérance au gluten alors que des praticiens ont perdu un temps infini à déterminer l’étiologie de la maladie cœliaque dont souffrait sa fille. Autant de témoignages de nature à convaincre de futurs clients !
Un accueil scientifique mitigé
Un premier article de Jane Kaye « The regulation of direct to consumer genetic tests » (Human Molecular Genetics, 2008), s’inquiète de la perte de contrôle des professionnels de santé, de l’évolution de la législation et de la confidentialité des données transitant par ces nouvelles sociétés de services. L’auteure appelle de ses vœux une régulation de cette « génomique récréative » où l’on peut partager sur les réseaux sociaux son potentiel génétique, ses risques de développer une maladie ou encore rechercher des « cousins » potentiels.
Deux ans après ce premier article, celui de Barbara Prainsack et Howard Wolinsky (Future Medicine, 2010) « Direct-to-consumer genome testing: opportunities for pharmacogenomics research ? » pondère le propos. Dans cet article, les auteurs ne s’inquiètent plus vraiment de l’existence d’un système divulguant des informations génomiques directement au consommateur sans passer par la case du prescripteur médical, mais s’interrogent sur les bénéfices que la recherche scientifique pourrait tirer de ces génotypages payés par les consommateurs eux-mêmes. Entrerait-on dans l’ère du libéralisme génomique ? L’article de Future Medicine développe l’aspect crowdsourcing (en français collaborat ou externalisation ouverte) en s’inquiétant légèrement tout de même d’un problème majeur : le respect de la vie privée…
Une inquiétude majeure
A une époque où des services étatiques développent des projets tels que PRISM, l’outil de surveillance du web utilisé par la NSA ou d’autres que la DGSE utiliserait, le consommateur de la génomique personnalisée pourrait et devrait s’inquiéter de la confidentialité des données génomiques le concernant. En effet, le web est le tuyau par lequel transite toutes les informations génomiques émanant des analyses de ces sociétés de service. La confidentialité des données semble être le nouveau frein au développement de ce type de marché.

Types d'altérations génomiques détectées par séquençage haut débit
Le séquençage massif parallélisé palliant les limites des anciennes méthodes, a depuis ces dernières années amorcé de nouvelles perspectives, comme l’étude d’altérations génomiques pour la compréhension de nombreuses maladies rares (Plan National Maladies Rares). De plus, la mise sur le marché des derniers séquenceurs de paillasse aux visées hospitalières (positionnement de marché de Ion torrent avec son PGM) contribue également au développement de nombreux diagnostics cliniques.
Face à cette révolution, il convient de faire face au goulot d’étranglement de l’analyse et de l’interprétation, en faisant appel à une stratégie plus appropriée que le séquençage de génome complet et qui consiste à focaliser les efforts de décodage sur les régions codantes enrichies (Exome, gènes cibles, etc…) par une étape préliminaire de d’enrichissement d’exons. Tout en contribuant à une diminution des coûts (on ne séquence que ce qui semble pertinent…), elle permet entre autre d’augmenter la profondeur de séquençage des régions d’intérêts, paramètre indispensable pour l’identification d’altérations génomiques.
Succinctement, les techniques d’enrichissement sont multiples et peuvent être regroupées en trois catégories sur la base de leur principe technique:
– Enrichissement par capture d’hybrides : La capture par hybridation des régions cibles est initiée à partir de la librairie de séquençage. Elle peut être effectuée soit en solution (billes magnétiques) soit sur support solide (microarrays). Récemment, de nombreuses solutions (commerciales ou « à façon ») ont été développées: SureSelect, Agilent – SeqCap EZ, Roche NimbleGen – Sequence Capture Arrays, Roche Nimblegen – FleXelect, FlexGen – MYselect, MYcroarray – TargetSeq, Life Technologies .
– Enrichissement par circularisation de fragments d’ADN : Cette méthode est supérieure à la précédente en terme de spécificité. En amont de la préparation de la librairie, les fragments d’ADNg (fragmentation enzymatique ou mécanique selon la méthode) sont enrichis via une sonde constituée d’une séquence universelle flanquée aux extrémités de séquences spécifiques de la région cible. HaloPlex, Agilent.
– Enrichissement par PCR : Cette approche intervient avant la préparation de la librairie et consiste grossièrement en une PCR multipléxée ciblant les régions d’intérêts. Une étape préliminaire de design des amorces est requise. SequalPrep, Life technologies. Tout juste disponible, Ion AmpliSeq Designer, Life Technologies propose un PCR Ultra-Plex (jusqu’à 1536 amplicons) avec un étape de digestion des amorces permettant de ne conserver que les régions d’intérêts lors du séquençage.
Une technologie récente et élégante faisant appel à la microfluidique et l’émulsion PCR permet l’amplification multipléxée (jusqu’à 20000) en micro-gouttelettes (une paire d’amorces par microréacteur) en un seul tube. Multiplicom, Fluidigm – RainDance.
Ceci étant, les considérations fondamentales dans l’analyse de variants résident notamment dans:
– la profondeur de séquençage
– l’homogénéité de couverture des régions d’intérêt
– la reproductibilité de la méthode
– la quantité d’ADN « Input » requis
– le nombre d’échantillons à traiter
– le coût global
L’étude récemment menée par Florian Mertes et al. (nov. 2011) souligne l’importance de ces notions et fait le postulat de performances de captures de régions cibles significativement différentes selon la méthode d’enrichissement employée.

- Comparaison des différentes techniques d’enrichissement de régions d’intérêts du gène PT53. La représentation supérieure illustre la profondeur de séquençage obtenue et la ligne en gras correspondante, la région utilisée pour la conception de sondes.

Il est impossible d’identifier une méthode d’enrichissement comme étant la meilleure notamment parce que ces approches sont en continuelle évolution et amélioration. Chacune d’elles répondent à des caractéristiques particulières et à des applications distinctes.
Si l’enrichissement par circularisation offre davantage de spécificité mais moins d’uniformité, l’inverse est aussi valable pour l’enrichissement par capture d’hybrides. Parmi les considérations dans le choix d’une méthode, le nombre d’échantillons et la taille des régions cibles sont essentiels. Si la capture d’hybrides est privilégiée pour l’analyse de mégabases (étude de l’exome) avec un nombre limité de cas, cette méthode sera délaissée au profit d’une approche PCR multipléxée pour l’étude d’un nombre restreint de régions cibles de petite taille appliquée à un grand nombre d’échantillons (Diagnostic).

L’association des méthodes d’enrichissement au séquençage haut débit offre des capacités technologiques ouvrant la voie vers de nombreuses perspectives. Toutefois, si l’émergence des séquenceurs de troisième génération s’accompagne à nouveau d’une diminution des coûts de séquençage, on est en droit de se demander si cette option restera aussi attractive.
 Noblegen Biosciences, start-up localisée dans le Massachusetts, ambitionne de commercialiser pour 2014 « optipore » (pour « optical detection » et « nanopore ») , un séquenceur de paillasse de troisième génération combinant nanotechnologies et un système de lecture optique permettant de réduire drastiquement les coûts de séquençage. L’objectif est de conquérir le marché des laboratoires cliniques dans la perspective de l’émergence d’une médecine personnalisée et de proposer ainsi un séquençage de génome humain complet à faible coût et dans un temps record. Dans cette course engagée, la société américaine se trouve en bonne position.
Noblegen Biosciences, start-up localisée dans le Massachusetts, ambitionne de commercialiser pour 2014 « optipore » (pour « optical detection » et « nanopore ») , un séquenceur de paillasse de troisième génération combinant nanotechnologies et un système de lecture optique permettant de réduire drastiquement les coûts de séquençage. L’objectif est de conquérir le marché des laboratoires cliniques dans la perspective de l’émergence d’une médecine personnalisée et de proposer ainsi un séquençage de génome humain complet à faible coût et dans un temps record. Dans cette course engagée, la société américaine se trouve en bonne position.
A l’instar du PGM Ion torrent, une puce en silicium constitue le coeur de la machine et renferme des centaines de nanopores.  La molécule d’ADN unique native destinée à être séquencée est tout d’abord convertie en une nouvelle molécule synthétique transformant chaque base par une séquence nucléotidique spécifique correspondante. A chacune des quatre signatures nucléotidiques correspond un « molecular beacon » complémentaire fluorescent initialement inactivé par un système de « quencher ». Leurs hybridations au brin néoformé aboutit à un ADN double brin. la molécule est dirigé vers les nanopores par des échanges ioniques et en raison de la taille des orifices, les « molecular beacon » sont contraints à se deshybrider libérant cette fois une fluorescence, lue par un capteur photographique de type CMOS, qui est traduite en séquence.
La molécule d’ADN unique native destinée à être séquencée est tout d’abord convertie en une nouvelle molécule synthétique transformant chaque base par une séquence nucléotidique spécifique correspondante. A chacune des quatre signatures nucléotidiques correspond un « molecular beacon » complémentaire fluorescent initialement inactivé par un système de « quencher ». Leurs hybridations au brin néoformé aboutit à un ADN double brin. la molécule est dirigé vers les nanopores par des échanges ioniques et en raison de la taille des orifices, les « molecular beacon » sont contraints à se deshybrider libérant cette fois une fluorescence, lue par un capteur photographique de type CMOS, qui est traduite en séquence.

L’ensemble des études et preuves de faisabilité sur lesquelles repose la technologie de séquençage « optipore » sont décrites et détaillées au travers de l’article libre d’accès ci dessous: A ce jour, peu de caractéristiques techniques liées à cette plateforme filtrent mais Frank Feist, cofondateur de la société, annonce une capacité de séquençage de 500 Gb/heure. De plus, en raison de l’étape de conversion initiale, la taille des fragments serait limitée à 200 bases.
A ce jour, peu de caractéristiques techniques liées à cette plateforme filtrent mais Frank Feist, cofondateur de la société, annonce une capacité de séquençage de 500 Gb/heure. De plus, en raison de l’étape de conversion initiale, la taille des fragments serait limitée à 200 bases.
Un soutien de 4,2 millions de dollars en septembre dernier de la part du National Human Genome Research Institute devrait permettre à la société américaine de conforter cette avancée prometteuse.
 L’étude de Fan et al publiée en 2008 et que nous avions évoquée au travers d’un article précédent, décrivait l’étude de faisabilité d’un diagnostic prénatal non invasif à partir de cellules d’origine fœtale provenant d’une simple prise de sang maternel.
L’étude de Fan et al publiée en 2008 et que nous avions évoquée au travers d’un article précédent, décrivait l’étude de faisabilité d’un diagnostic prénatal non invasif à partir de cellules d’origine fœtale provenant d’une simple prise de sang maternel.
Ces travaux ont fait l’objet de controverses et même si la détection de la trisomie 21 est rendue possible , cela ne l’ était déjà plus pour les trisomies 18 ou 13, par exemple. En cause, la méthode d’analyse employée : l’ensemble des séquences sont alignées par rapport à un génome de référence et l’aneuploïdie foetale est détectée par une surreprésentation du chromosome correspondant, au sein du jeu de données. La sensibilité et l’efficacité de la méthode sont ici directement liées à la profondeur de séquençage. Cette approche ne permet pas de prendre en compte les biais aléatoires ou systématiques liés aux techniques de séquençage, facteurs primordiaux pour cette application.
Dernièrement, les entreprises Sequenom et Verinata Health ont publié des travaux (Sehnert et al.,2011) portant sur le dépistage d’aneuploïdies via le séquençage à haut débit, à partir de cellules d’origine fœtale issues d’une prise de sang maternel : 100% des cas de trisomies 21, 18 et autres anomalies ont été décelés ( soit 27 caryotypes anormaux parmi 48 patients ).

Cette efficacité repose sur une optimisation de l’analyse bioinformatique et notamment sur une considération pour les variations intra et inter-run qui se situent bien souvent à la frontière avec les faibles modifications de la distribution des séquences entre un cas sain et un cas d’aneuploïdie. L’algorithme développé utilise des valeurs de chromosome normalisées établies sur la base d’un ensemble de données de séquençages provenant d’échantillons parmi lesquels certains sont connus comme ayant un caryotype anormal.
En podcast de « clinical chemistry », le Dr. Richard P. Rava, co-auteur de l’article proposé précédemment, revient sur des points de détails et explications concernant le développement de l’algorithme optimisé pour les détections d’aneuploïdies.
L’effervescence médiatique liée au séquençage de la bactérie Escherichia coli responsable de l’intoxication d’une dizaine de personnes dans le Nord de la France (juin 2011), a engendré de multiples reportages et communiqués. Parmi eux, celui de l’AFP, et ce paragraphe comme point de départ à cet article :
« L’Institut est équipé depuis un mois d’un séquenceur « à haut débit » qui permet de déchiffrer un génome en un temps record à partir d’une « librairie » d’ADN stockée sur une sorte de puce électronique. Cette technologie est notamment utilisée dans la recherche sur le cancer et pourrait remplacer dans un futur proche l’amniosynthèse (sic). »
En France, deux méthodes « invasives » permettent de réaliser un diagnostic prénatal : L’amniocentèse ou prélèvement du liquide amniotique, et une biopsie de trophoblastes ou prélèvement des cellules du futur placenta.
Toutefois, cet acte cause chaque année environ 1% de fausses couches. Rapporté aux 90000 amniocentèses par an, on déplore jusqu’à 900 pertes de fœtus, qui paradoxalement sont sain dans la majorité des cas.
Ces conséquences iatrogènes sont terribles sur le plan humain et il apparait évident de mettre au point une méthode beaucoup moins dangereuse pour la mère et surtout pour le fœtus.
Depuis plusieurs années, il a été démontré la présence de cellules d’origine fœtales dans le sang maternel dès la huitième semaine de grossesse. Cette particularité a dernièrement fait quelques émules et ont décidé de l’exploiter pour la mise au point d’un diagnostic prénatal « non invasif ». Le point critique repose sur la très faible proportion de ces cellules provenant du fœtus ; de l’ordre d’une pour dix millions de globules blancs et cinq milliards de glo¬bules rouges par millilitre de sang maternelle.
L’une des méthodes les plus encourageantes, réalisée par l’équipe de Patrizia Paterlini-Bréchot, Unité Inserm U807, repose sur une méthode d’enrichissement par la taille des cellules trophoblastique dans le sang, dite « ISET » ( Isolation by Size of Epithelial Tumor / Trophoblastic cells ), leur caractérisation par la technique des empreintes génétiques, puis séquençage de l’ADN extrait. La société « Rarecells » a par ailleurs était créée sur cette base.
Cette méthode sensible et spécifique à 100%, a déjà été validée techniquement et cliniquement pour le diagnostic prénatal non invasif de l’amyotrophie spinale (Beroud C., Karliova M., Bonnefont J.P., et al. Prenatal diagnosis of spinal muscular atrophy by genetic analysis of circulating fetal cells Lancet 2003 ; 361 (9362) : 1013-1014) et de la mucoviscidose ( « Recherche de la mutation du gène CFTR » Saker A., Benachi A., Bonnefont J.P., et al. Genetic characterisation of circulating fetal cells allows non-invasive prenatal diagnosis of cystic fibrosis Prenat Diagn 2006 ; 26 (10) : 906-916) , et techniquement seulement pour la trisomie 21.
Une seconde approche, réalisée par des chercheurs de l’Université de Stanford, est basée sur l’étude de l’ADN fœtal libre extrait du sang maternel puis analysé grâce aux séquenceurs à haut débit.
Dans le cas de la trisomie 21 par exemple, l’assignement de plusieurs dizaines de milliers de séquences présenterait un déséquilibre statistique même si la proportion d’ADN fœtal par rapport à l’ADN maternel est extrêmement faible (Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proceedings of the National Science Academy of the USA, 2008, 105, 16266–71). Si cette approche semble très attractive, plusieurs interrogations se posent suite à la lecture du travail scientifique, à la fois sur la méthodologie, l’analyse statistique et l’évaluation des résultats. (Commentaires sur les études de Fan et al.)
Dans l’éventualité d’un diagnostic basé sur le séquençage complet d’un génome, ou davantage ciblé tel l’ « amplicon sequencing » des gènes d’intérêts, la méthode non invasive s’appuyant sur une analyse globale du sang maternel nécessiterait une profondeur de séquençage conséquente. Ceci aurait pour incidence une orientation technique tournée vers des appareils de type « Solid 5500 » ou encore « Illumina HiSeq 2000 », cracheurs de « reads ».
Dans le cadre d’une application diagnostique, l’investissement financier est l’un des aspects considéré et en ce sens la méthode « ISET », plus contraignante dans la préparation du matériel biologique, permets néanmoins d’accéder, dans la perspective d’un séquençage ciblé, aux séquenceurs de paillasse, cinq à dix fois moins cher à l’achat et à des coûts de « runs » inégalés à ce jour. Record actuellement détenu par le PGM, Ion torrent dont la capacité des puces actuelles de 100Mb et d’1Gb à venir d’ici la fin d’année 2011, permettrait un multiplexage soit d’individus pour une mutation donnée, ou encore, imaginer une liste de séquences d’intérêts à diagnostiquer par individu.
Il y a fort à parier que le séquençage à haut débit finisse par devenir un outil incontournable dans le monde du diagnostic prénatal d’autant plus que les coûts d’équipement et de fonctionnement ne cessent de diminuer. Dans cette perspective, l’évolution vers les séquenceurs haut-débit de 3ème génération (à partir de cellule unique) pourrait faire la part belle à l’utilisation des cellules trophoblastiques.
Au travers de ces avancées et de l’ère du séquençage à haut débit, les perspectives d’un diagnostic prénatal non invasif semblent se dessiner et laisse entrevoir un énorme soulagement. Aussi, la possibilité de mettre en place des plates-formes permettant de cribler l’essentiel des pathologies génétiques est envisagée.
Aussitôt, les détracteurs soulèvent la notion d’eugénisme, dénonçant un diagnostic prénatal intégral en vue d’une « traque » des individus « non conformes ».
Il apparait évident que ces avancées technologiques risquent très prochainement de soulever de réels débats de société…
Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.


{kind=link}
{kind=link}
{kind=link}