
Posts by: renaud blervaque
Un document qui n’est pas sans rappeler celui publié en novembre 2011, en intégrant les dernières mises à jour.
Cet arbre de décision est l’occasion de retrouver l’ensemble des modalités de séquençage qu’offre la technologie Ion Torrent via le PGM. En attendant l’arrivée du kit permettant de séquencer 400b, l’essentiel des modifications résident dans la préparation des échantillons avec notamment de nouvelles indexations (évolution du kit à 96 barcodes en DNA-seq et apparition du kit à 16 barcodes en RNA-seq), un élargissement du panel de primers dédiés à la recherche sur le cancer, etc…
Pour les privilégiées de la Ion Community, ce document est également l’occasion de retrouver aisément la liste des protocoles Ion Torrent selon l’ application d’intérêt en cliquant sur le lien en bas de page.


Face aux arguments commerciaux développés en faveur de tels ou tels séquenceurs de paillasse à haut débit, l’étude menée par Loman et al., Nature Biotechnology propose une comparaison des performances du 454 GS Junior (Roche), du MiSeq (Illumina) et du PGM Ion torrent (Life Technologies), à partir du séquençage de novo d’un isolat d’E.coli O104 :H4 responsable de l’intoxication alimentaire en Allemagne (Mai 2011). Pour cela, le génome de référence a été généré à l’aide du GS FLX (Roche) et permettra d’évaluer l’efficacité des séquenceurs et algorithmes d’assemblages associés.
Sur la base des caractéristiques techniques établies en 2011, les trois plateformes ont permis de générer un draft exploitable du génome bactérien sans pour autant parvenir à le reconstituer à 100%. Par conséquent, il est impossible de désigner un séquenceur comme étant celui de choix, chaque plateforme étant un compromis entre avantages et inconvénients.
Deux « runs » de 454 GS Junior (associés au bénéfice de reads de 600 bases) ont permis d’atteindre 96,28% de couverture par rapport au génome de référence (Contre 96,05% pour le MiSeq et 95,4% pour le PGM Ion torrent).
Malgré une capacité de séquençage du PGM Ion torrent supérieure à celle du 454 GS Junior (100Mb contre 70Mb) et une précision de séquençage voisine (Q20, soit 1 erreur toutes les 100 bases), la technologie reposant sur les semiconducteurs fait face à ses limites au niveau de la gestion des homopolymères et accessoirement d’une longueur de reads de 100 bases.
Vingt fois supérieur au 454 GS Junior en terme de quantité de bases décodées, un « run » de MiSeq ne parvient qu’à talonner les performances d’un double « run » de la technologie Roche. Bien qu’étant la seule à gérer les homopolymères et la seule doter d’une précision à Q30 (soit 1 erreur toutes les 1000 bases), la technologie Illumina faillit sur ses tailles de « reads » qui ne permettent pas une gestion optimum des séquences répétées lors de l’assemblage.
L’approche analytique est également évoquée et selon le système d’assemblage utilisé (Velvet, CLC, MIRA), de nombreuses différences sont observées.
Enfin, ce document est l’occasion de rassembler des notions assez pragmatiques comme le tarif des plateformes (et annexes associées), la durée d’un « run » de séquençage ou encore la quantité de données générées.
Ceci étant, on est en droit d’émettre quelques réticences quant à la pertinence de cette étude qui base son comparatif sur une application (séquençage de novo) qui n’est pas la vocation première de cette catégorie de séquenceurs. Par ailleurs, dans le laps de temps où cette étude a été réalisée et la parution des résultats, la technologique du PGM Ion torrent a évolué. Testé sur les bases d’une puce 316 (100Mb) et d’une technologie de 100 bases en longueur de « reads », le PGM bénéficie désormais de la puce 318 (1 Gb) associée à une technologie de reads de 200 bases. Sans tarder, Life technologies réagit (“Loman et al reflects the past, not the present”) et propose une mise à jour au travers de ce document.


Types d'altérations génomiques détectées par séquençage haut débit
Le séquençage massif parallélisé palliant les limites des anciennes méthodes, a depuis ces dernières années amorcé de nouvelles perspectives, comme l’étude d’altérations génomiques pour la compréhension de nombreuses maladies rares (Plan National Maladies Rares). De plus, la mise sur le marché des derniers séquenceurs de paillasse aux visées hospitalières (positionnement de marché de Ion torrent avec son PGM) contribue également au développement de nombreux diagnostics cliniques.
Face à cette révolution, il convient de faire face au goulot d’étranglement de l’analyse et de l’interprétation, en faisant appel à une stratégie plus appropriée que le séquençage de génome complet et qui consiste à focaliser les efforts de décodage sur les régions codantes enrichies (Exome, gènes cibles, etc…) par une étape préliminaire de d’enrichissement d’exons. Tout en contribuant à une diminution des coûts (on ne séquence que ce qui semble pertinent…), elle permet entre autre d’augmenter la profondeur de séquençage des régions d’intérêts, paramètre indispensable pour l’identification d’altérations génomiques.
Succinctement, les techniques d’enrichissement sont multiples et peuvent être regroupées en trois catégories sur la base de leur principe technique:
– Enrichissement par capture d’hybrides : La capture par hybridation des régions cibles est initiée à partir de la librairie de séquençage. Elle peut être effectuée soit en solution (billes magnétiques) soit sur support solide (microarrays). Récemment, de nombreuses solutions (commerciales ou « à façon ») ont été développées: SureSelect, Agilent – SeqCap EZ, Roche NimbleGen – Sequence Capture Arrays, Roche Nimblegen – FleXelect, FlexGen – MYselect, MYcroarray – TargetSeq, Life Technologies .
– Enrichissement par circularisation de fragments d’ADN : Cette méthode est supérieure à la précédente en terme de spécificité. En amont de la préparation de la librairie, les fragments d’ADNg (fragmentation enzymatique ou mécanique selon la méthode) sont enrichis via une sonde constituée d’une séquence universelle flanquée aux extrémités de séquences spécifiques de la région cible. HaloPlex, Agilent.
– Enrichissement par PCR : Cette approche intervient avant la préparation de la librairie et consiste grossièrement en une PCR multipléxée ciblant les régions d’intérêts. Une étape préliminaire de design des amorces est requise. SequalPrep, Life technologies. Tout juste disponible, Ion AmpliSeq Designer, Life Technologies propose un PCR Ultra-Plex (jusqu’à 1536 amplicons) avec un étape de digestion des amorces permettant de ne conserver que les régions d’intérêts lors du séquençage.
Une technologie récente et élégante faisant appel à la microfluidique et l’émulsion PCR permet l’amplification multipléxée (jusqu’à 20000) en micro-gouttelettes (une paire d’amorces par microréacteur) en un seul tube. Multiplicom, Fluidigm – RainDance.
Ceci étant, les considérations fondamentales dans l’analyse de variants résident notamment dans:
– la profondeur de séquençage
– l’homogénéité de couverture des régions d’intérêt
– la reproductibilité de la méthode
– la quantité d’ADN « Input » requis
– le nombre d’échantillons à traiter
– le coût global
L’étude récemment menée par Florian Mertes et al. (nov. 2011) souligne l’importance de ces notions et fait le postulat de performances de captures de régions cibles significativement différentes selon la méthode d’enrichissement employée.

- Comparaison des différentes techniques d’enrichissement de régions d’intérêts du gène PT53. La représentation supérieure illustre la profondeur de séquençage obtenue et la ligne en gras correspondante, la région utilisée pour la conception de sondes.

Il est impossible d’identifier une méthode d’enrichissement comme étant la meilleure notamment parce que ces approches sont en continuelle évolution et amélioration. Chacune d’elles répondent à des caractéristiques particulières et à des applications distinctes.
Si l’enrichissement par circularisation offre davantage de spécificité mais moins d’uniformité, l’inverse est aussi valable pour l’enrichissement par capture d’hybrides. Parmi les considérations dans le choix d’une méthode, le nombre d’échantillons et la taille des régions cibles sont essentiels. Si la capture d’hybrides est privilégiée pour l’analyse de mégabases (étude de l’exome) avec un nombre limité de cas, cette méthode sera délaissée au profit d’une approche PCR multipléxée pour l’étude d’un nombre restreint de régions cibles de petite taille appliquée à un grand nombre d’échantillons (Diagnostic).

L’association des méthodes d’enrichissement au séquençage haut débit offre des capacités technologiques ouvrant la voie vers de nombreuses perspectives. Toutefois, si l’émergence des séquenceurs de troisième génération s’accompagne à nouveau d’une diminution des coûts de séquençage, on est en droit de se demander si cette option restera aussi attractive.
 Noblegen Biosciences, start-up localisée dans le Massachusetts, ambitionne de commercialiser pour 2014 « optipore » (pour « optical detection » et « nanopore ») , un séquenceur de paillasse de troisième génération combinant nanotechnologies et un système de lecture optique permettant de réduire drastiquement les coûts de séquençage. L’objectif est de conquérir le marché des laboratoires cliniques dans la perspective de l’émergence d’une médecine personnalisée et de proposer ainsi un séquençage de génome humain complet à faible coût et dans un temps record. Dans cette course engagée, la société américaine se trouve en bonne position.
Noblegen Biosciences, start-up localisée dans le Massachusetts, ambitionne de commercialiser pour 2014 « optipore » (pour « optical detection » et « nanopore ») , un séquenceur de paillasse de troisième génération combinant nanotechnologies et un système de lecture optique permettant de réduire drastiquement les coûts de séquençage. L’objectif est de conquérir le marché des laboratoires cliniques dans la perspective de l’émergence d’une médecine personnalisée et de proposer ainsi un séquençage de génome humain complet à faible coût et dans un temps record. Dans cette course engagée, la société américaine se trouve en bonne position.
A l’instar du PGM Ion torrent, une puce en silicium constitue le coeur de la machine et renferme des centaines de nanopores.  La molécule d’ADN unique native destinée à être séquencée est tout d’abord convertie en une nouvelle molécule synthétique transformant chaque base par une séquence nucléotidique spécifique correspondante. A chacune des quatre signatures nucléotidiques correspond un « molecular beacon » complémentaire fluorescent initialement inactivé par un système de « quencher ». Leurs hybridations au brin néoformé aboutit à un ADN double brin. la molécule est dirigé vers les nanopores par des échanges ioniques et en raison de la taille des orifices, les « molecular beacon » sont contraints à se deshybrider libérant cette fois une fluorescence, lue par un capteur photographique de type CMOS, qui est traduite en séquence.
La molécule d’ADN unique native destinée à être séquencée est tout d’abord convertie en une nouvelle molécule synthétique transformant chaque base par une séquence nucléotidique spécifique correspondante. A chacune des quatre signatures nucléotidiques correspond un « molecular beacon » complémentaire fluorescent initialement inactivé par un système de « quencher ». Leurs hybridations au brin néoformé aboutit à un ADN double brin. la molécule est dirigé vers les nanopores par des échanges ioniques et en raison de la taille des orifices, les « molecular beacon » sont contraints à se deshybrider libérant cette fois une fluorescence, lue par un capteur photographique de type CMOS, qui est traduite en séquence.

L’ensemble des études et preuves de faisabilité sur lesquelles repose la technologie de séquençage « optipore » sont décrites et détaillées au travers de l’article libre d’accès ci dessous: A ce jour, peu de caractéristiques techniques liées à cette plateforme filtrent mais Frank Feist, cofondateur de la société, annonce une capacité de séquençage de 500 Gb/heure. De plus, en raison de l’étape de conversion initiale, la taille des fragments serait limitée à 200 bases.
A ce jour, peu de caractéristiques techniques liées à cette plateforme filtrent mais Frank Feist, cofondateur de la société, annonce une capacité de séquençage de 500 Gb/heure. De plus, en raison de l’étape de conversion initiale, la taille des fragments serait limitée à 200 bases.
Un soutien de 4,2 millions de dollars en septembre dernier de la part du National Human Genome Research Institute devrait permettre à la société américaine de conforter cette avancée prometteuse.
Ce post fait naturellement suite à celui dédié à la seconde génération de séquenceurs multi-parallélisés, et conserve la même approche, à savoir un tour d’horizon des technologies et une évocations des informations générales sur le sujet.
A l’instar du PGM de Ion torrent mis sur le marché depuis un an (10Mb – reads 100b – 06.2011 / 100Mb – reads 200b -11.2011 / 1Gb – reads 400b – prévu début 2012), la seconde génération de séquenceurs haut débit tend vers une production de reads de plus en plus longs et de moins en moins chère. Toutefois, on est en droit de se demander quelle sera leur pérennité face à la 3éme génération répondant à un cahier des charges assez similaire et la possibilité de bénéficier de nouvelles applications.

Le principe de la 3ème génération peut être symbolisé par le séquençage d’une molécule d’ADN sans étape de pré-amplification (contrairement à la génération actuelle type 454 Roche, SOLiD Life technologie, Ion Proton, PGM Ion torrent, HiSeq Illumina, …) en conservant l’incorporation de nucléotides, par cycles ou non ( dans ce dernier cas, le terme de « Séquençage d’ADN simple molécule en temps réel » est approprié).
Les technologies « SMS » pour « Single Molecule Sequencing » peuvent être regroupées selon trois catégories:
– Technologies de séquençage en temps réel impliquant la synthèse du brin d’ADN complémentaire via une ADN polymérase.
– Technologies de séquençage par détection des bases successives d’une molécule d’ADN au travers de nanopores.
– Technologies de séquençage basées sur des techniques de microscopie.
En combinant les dernières avancées dans la nanofabrication, la chimie de surface et l’optique, Pacific Biosciences (Pacbio RS) a lancé une plateforme technologique puissante appelée technologie de molécule unique en temps réel, ou « SMRT » pour « Single Molecule Real-time sequencing ». Parmi ses concurrents directs, Helicos Biosciences (Helicos) qualifié « tSMS » pour « True Single Molecule Sequencing ». Malgré le recours à une technologie analogue, la mention « Temps réel » auquel il échappe est simplement liée à une incorporation cyclique des nucléotides fluorescents.
D’autres technologies, à des degrés de développement plus ou moins avancé, sont dans les tuyaux et qui sait de Noblegen, Starlight, Cracker Bio, NABSys, Halcyon, ou autres… révolutionnera encore un peu plus cet univers du haut débit et suivra le chemin emprunté dernièrement par Oxford Nanopore …


Ion Proton Sequencer
Dernier né de la gamme des séquenceurs de paillasse, le « Ion Proton Sequencer » (149.000 $) est le premier de sa catégorie à permettre le séquençage en quelques heures du génome humain et à faible coût (1.000 $). A l’instar du PGM Ion Torrent, cette nouvelle plateforme est basée sur la technologie des semi-conducteurs (et la détection de la libération d’ions H+ suite à la polymérisation de dNTP).
La puce « Ion Proton ™ I »,disponible mi-2012, aura une capacité de séquençage 1000 fois supérieure au premier format de puce 314 du PGM Ion torrent soit 10 Gb (davantage orientée pour le séquençage d’exome humain). Le second format (puce « Ion Proton ™ II ») , disponible environ six mois plus tard aura une capacité de 100 Gb (Séquençage Génome humain).
Compte tenu de l’apparente similarité entre les deux plateformes de chez Life technologies, l’évolution réside essentiellement dans l’architecture de la puce avec une augmentation du nombre de puits ( 165 millions de puits pour la puce « Ion Proton ™ I » et 660 millions pour la puce « Ion Proton ™ II » soit respectivement 100 et 1000 fois plus que la puce 314 du PGM ), l’une des bases de la capacité de séquençage (en complément de la longueur des reads générés).
Le terme « Nouvelle génération de séquençage à haut-débit » ( ou « Next generation sequencing » ) regroupe l’ensemble des technologies ou plateformes de séquençage développées depuis 2005 par quelques sociétés de biotechnologies.
L’objectif de cet article est de proposer de manière synthétique, un tour d’horizon des différents principes et caractéristiques de ces nouveaux outils et ainsi fournir quelques orientations et solutions techniques en réponses à des questions biologiques.
La position actuelle dans laquelle nous nous trouvons, entre la commercialisation de certains séquenceurs et ceux en cours de développement, est caractéristique d’une période charnière dissociant les technologies à haut débit dites de 2ème génération qui requièrent une étape d’amplification des molécules d’ADN en amont du décodage, de celles dites de 3ème génération permettant le décryptage direct d’une seule molécule d’ADN. Cette dernière catégorie fera à elle seule l’objet d’un prochain article.
Le marché des séquenceurs de 2éme génération est couvert par 3 grand groupes que sont Roche, Illumina et Life Technologies ayant respectivement proposés de manière successive, leur première plateforme à savoir le 454, le Genome Analyser et enfin le SOLiD. Depuis, le marché s’est étoffé proposant un panel de technologies au principe et caractéristiques propres telles qu’elles sont mentionnées ci dessous. A noter que parmi ce panel, le PGM, Ion torrent est le seul a connaitre une évolution constante en terme de capacités de séquençage (10Mb – reads de 100b – Juin 2011 / 100Mb – reads de 100b – Sept 2011 / 100Mb – reads 200b – Nov 2011 / 1Gb – Jan 2012 )

Chaque plateforme possède ses avantages et inconvénients et nombreuses sont celles configurées pour répondre à de nombreuses approches « omics », dans certaines limites. Il s’agira de faire un choix technologique selon les champs d’applications souhaités.
De manière générale, le type d’organisme étudié prédéterminera la technologie à employer. La notion de profondeur est récurrente à chaque application et dans l’objectif d’un reséquençage, le choix de la plateforme peut être identifié, de manière simplifiée, sur la base d’un calcul rapide ( P = N / L où P: Profondeur, N: Nombre des nucléotides totaux des reads, L: Taille du génome étudié).
Concernant les séquenceurs de 2ème génération, le séquençage de novo est une application mentionnée chez de nombreux fournisseurs (cf le tableau ci-dessous). Toutefois, l’association de deux technologies générant à la fois des reads longs (type 454, Roche) et une profondeur conséquente (type GAIIx, Illumina) palliant aux problèmes liés aux homopolymères et erreurs de séquençage, est préconisée (Au cours de l’article à venir sur les séquenceurs de 3ème génération, nous aborderons les plateformes davantage configurées pour cette application).
Ce paramètre de profondeur sera également à prendre en considération pour les champs d’applications incluant la notion d’analyse quantitative (RNAseq, ChIPseq, …). Si la profondeur permet d’atténuer les erreurs de séquençage, il reste néanmoins préférable de s’orienter vers des technologies à Q30 minimum (1 erreur sur 1000) pour la détection de SNPs.

Selon les technologies évoquées ci-dessus, les caractéristiques et champs d’applications ont évolués. Aussi, je vous propose de retrouver l’ensemble de ces informations actualisées en cliquant sur ce lien.
L’ensemble des informations sont détaillées dans l’article mentionné ci-dessous:

 L’étude de Fan et al publiée en 2008 et que nous avions évoquée au travers d’un article précédent, décrivait l’étude de faisabilité d’un diagnostic prénatal non invasif à partir de cellules d’origine fœtale provenant d’une simple prise de sang maternel.
L’étude de Fan et al publiée en 2008 et que nous avions évoquée au travers d’un article précédent, décrivait l’étude de faisabilité d’un diagnostic prénatal non invasif à partir de cellules d’origine fœtale provenant d’une simple prise de sang maternel.
Ces travaux ont fait l’objet de controverses et même si la détection de la trisomie 21 est rendue possible , cela ne l’ était déjà plus pour les trisomies 18 ou 13, par exemple. En cause, la méthode d’analyse employée : l’ensemble des séquences sont alignées par rapport à un génome de référence et l’aneuploïdie foetale est détectée par une surreprésentation du chromosome correspondant, au sein du jeu de données. La sensibilité et l’efficacité de la méthode sont ici directement liées à la profondeur de séquençage. Cette approche ne permet pas de prendre en compte les biais aléatoires ou systématiques liés aux techniques de séquençage, facteurs primordiaux pour cette application.
Dernièrement, les entreprises Sequenom et Verinata Health ont publié des travaux (Sehnert et al.,2011) portant sur le dépistage d’aneuploïdies via le séquençage à haut débit, à partir de cellules d’origine fœtale issues d’une prise de sang maternel : 100% des cas de trisomies 21, 18 et autres anomalies ont été décelés ( soit 27 caryotypes anormaux parmi 48 patients ).

Cette efficacité repose sur une optimisation de l’analyse bioinformatique et notamment sur une considération pour les variations intra et inter-run qui se situent bien souvent à la frontière avec les faibles modifications de la distribution des séquences entre un cas sain et un cas d’aneuploïdie. L’algorithme développé utilise des valeurs de chromosome normalisées établies sur la base d’un ensemble de données de séquençages provenant d’échantillons parmi lesquels certains sont connus comme ayant un caryotype anormal.
En podcast de « clinical chemistry », le Dr. Richard P. Rava, co-auteur de l’article proposé précédemment, revient sur des points de détails et explications concernant le développement de l’algorithme optimisé pour les détections d’aneuploïdies.
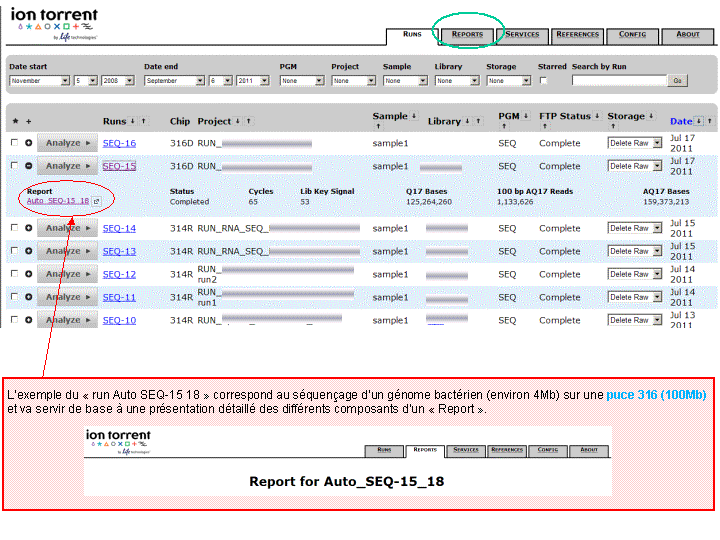
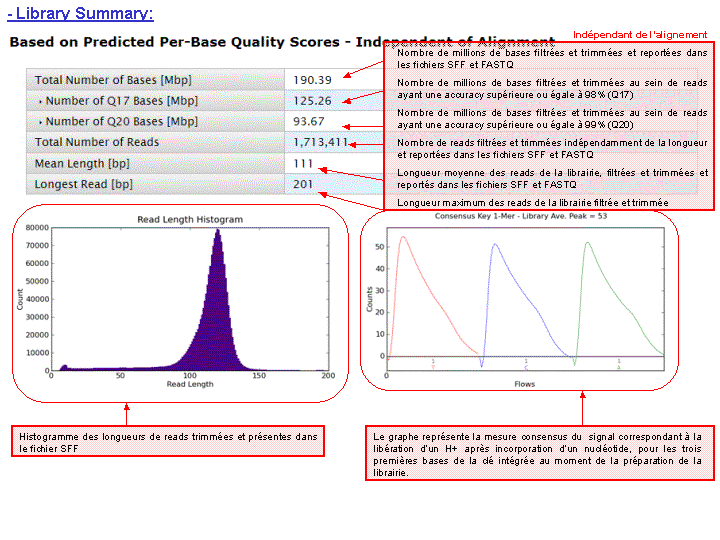
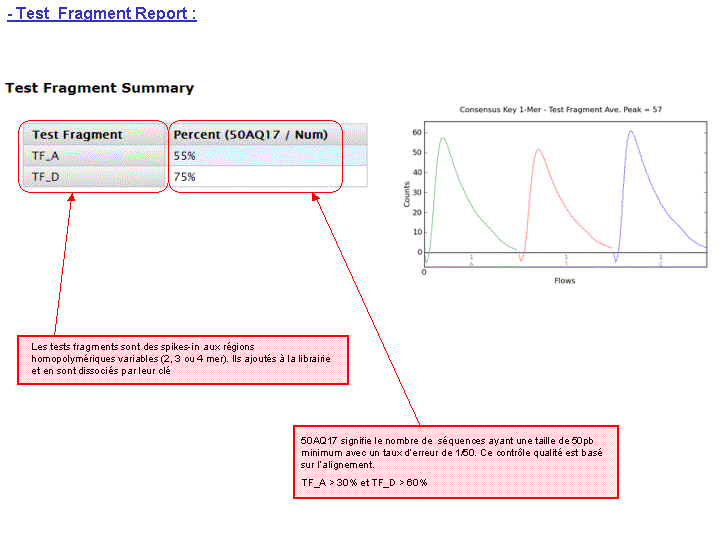
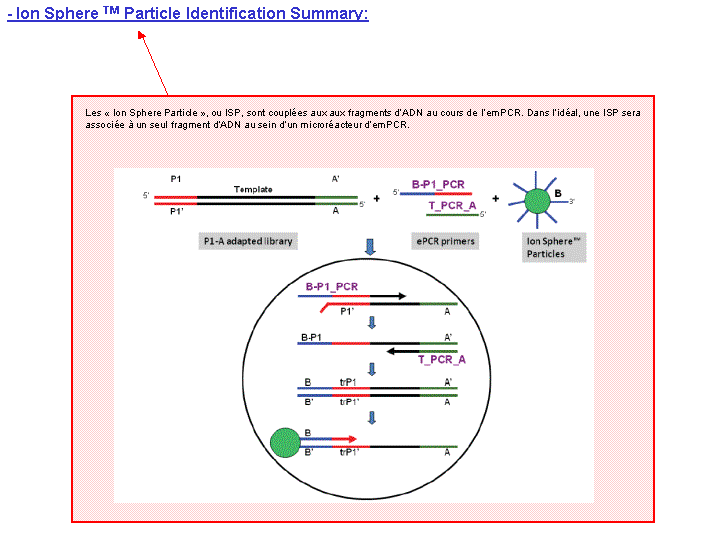
A l’issue d’un « run » de séquençage Ion Torrent (PGM), l’ensemble du signal brut (ionogramme) est converti en séquences et stocké au niveau du serveur. Pour chaque « run de re-séquençage», un alignement préliminaire est réalisé sur la base du génome de référence mentionné lors de l’initialisation du PGM. Cette information est reprise au travers d’un rapport qui comporte également un ensemble de paramètres, que l’on se propose de détailler ci-dessous :
(Cliquer pour agrandir) 
Le rapport se divise en 5 sections:

")

")
")





L ’évolution de la technologie Ion torrent permet depuis ces derniers jours, d’accéder au format de puce « 316 » succédant à la « 314 ». Ces puces multiplient par 10 la quantité de données générées à l’issue d’un « run » de séquençage aboutissant ainsi à 100 Mb.
De manière à bénéficier de la capacité totale de ces puces, une mise à jour du serveur, ainsi qu’une configuration du PGM en version 1.4 sont nécessaires et ont été réalisées ce jour. Ces modifications se traduisent par une amélioration de l’algorithme d’analyse (basecalling, alignement, …) optimisant le nombre de reads considérées et traitées. Le développement apporté au niveau des « runs » permet désormais d’intégrer des « reads » de plus grande taille (jusqu’à 200b environ) directement lié à une amélioration de la précision de séquençage.

Impact de la version1.4 à travers le report d'un "run"
Techniquement, quelques modifications au niveau du PGM ont conduit au remplacement de certaines tubulures (diamètre supérieur aux précédentes) : Cet épisode fut l’occasion de découvrir les entrailles de la machine…


Qui sommes nous?
Christophe Audebert [@]
 En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
En charge de la plateforme génomique
du département recherche et développement
de la société Gènes Diffusion .
Renaud Blervaque [@]
 Biologiste moléculaire, chargé d'études génomiques.
Biologiste moléculaire, chargé d'études génomiques.
Gaël Even [@]
 Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.
Responsable bioinformatique au sein
du département recherche et développement de la société Gènes Diffusion.


{kind=link}
{kind=link}
{kind=link}
{kind=link}